In genomic fields, it’s very common to explore the gene expression profile of one or a list of genes involved in a pathway of interest. Here, we present some helper functions in the ggpubr R package to facilitate exploratory data analysis (EDA) for life scientists.
Clik here to view.
Exploratory Data visualization: Gene Expression Data
Standard graphical techniques used in EDA, include:
- Box plot
- Violin plot
- Stripchart
- Dot plot
- Histogram and density plots
- ECDF plot
- Q-Q plot
All these plots can be created using the ggplot2 R package, which is highly flexible.
However, to customize a ggplot, the syntax might appear opaque for a beginner and this raises the level of difficulty for researchers with no advanced R programming skills. If you’re not familiar with ggplot2 system, you can start by reading our Guide to Create Beautiful Graphics in R.
Previously, we described how to Add P-values and Significance Levels to ggplots. In this article, we present the ggpubr package, a wrapper around ggplot2, which provides some easy-to-use functions for creating ‘ggplot2’- based publication ready plots. We’ll use the ggpubr functions to visualize gene expression profile from TCGA genomic data sets.
Contents:
Prerequisites
ggpubr package
Required R package: ggpubr (version >= 0.1.3).
- Install from CRAN as follow:
install.packages("ggpubr")- Or, install the latest developmental version from GitHub as follow:
if(!require(devtools)) install.packages("devtools")
devtools::install_github("kassambara/ggpubr")- Load ggpubr:
library(ggpubr)TCGA data
The Cancer Genome Atlas (TCGA) data is a publicly available data containing clinical and genomic data across 33 cancer types. These data include gene expression, CNV profiling, SNP genotyping, DNA methylation, miRNA profiling, exome sequencing, and other types of data.
The RTCGA R package, by Marcin Marcin Kosinski et al., provides a convenient solution to access to clinical and genomic data available in TCGA. Each of the data packages is a separate package, and must be installed (once) individually.
The following R code installs the core RTCGA package as well as the clinical and mRNA gene expression data packages.
# Load the bioconductor installer.
source("https://bioconductor.org/biocLite.R")
# Install the main RTCGA package
biocLite("RTCGA")
# Install the clinical and mRNA gene expression data packages
biocLite("RTCGA.clinical")
biocLite("RTCGA.mRNA")To see the type of data available for each cancer type, use this:
library(RTCGA)
infoTCGA()# A tibble: 38 x 13
Cohort BCR Clinical CN LowP Methylation mRNA mRNASeq miR miRSeq RPPA MAF rawMAF
*
1 ACC 92 92 90 0 80 0 79 0 80 46 90 0
2 BLCA 412 412 410 112 412 0 408 0 409 344 130 395
3 BRCA 1098 1097 1089 19 1097 526 1093 0 1078 887 977 0
4 CESC 307 307 295 50 307 0 304 0 307 173 194 0
5 CHOL 51 45 36 0 36 0 36 0 36 30 35 0
6 COAD 460 458 451 69 457 153 457 0 406 360 154 367
7 COADREAD 631 629 616 104 622 222 623 0 549 491 223 489
8 DLBC 58 48 48 0 48 0 48 0 47 33 48 0
9 ESCA 185 185 184 51 185 0 184 0 184 126 185 0
10 FPPP 38 38 0 0 0 0 0 0 23 0 0 0
# ... with 28 more rows More information about the disease names can be found at: http://gdac.broadinstitute.org/
Gene expression data
The R function expressionsTCGA() [in RTCGA package] can be used to easily extract the expression values of genes of interest in one or multiple cancer types.
In the following R code, we start by extracting the mRNA expression for five genes of interest - GATA3, PTEN, XBP1, ESR1 and MUC1 - from 3 different data sets:
- Breast invasive carcinoma (BRCA),
- Ovarian serous cystadenocarcinoma (OV) and
- Lung squamous cell carcinoma (LUSC)
library(RTCGA)
library(RTCGA.mRNA)
expr <- expressionsTCGA(BRCA.mRNA, OV.mRNA, LUSC.mRNA,
extract.cols = c("GATA3", "PTEN", "XBP1","ESR1", "MUC1"))
expr# A tibble: 1,305 x 7
bcr_patient_barcode dataset GATA3 PTEN XBP1 ESR1 MUC1
1 TCGA-A1-A0SD-01A-11R-A115-07 BRCA.mRNA 2.870500 1.3613571 2.983333 3.0842500 1.652125
2 TCGA-A1-A0SE-01A-11R-A084-07 BRCA.mRNA 2.166250 0.4283571 2.550833 2.3860000 3.080250
3 TCGA-A1-A0SH-01A-11R-A084-07 BRCA.mRNA 1.323500 1.3056429 3.020417 0.7912500 2.985250
4 TCGA-A1-A0SJ-01A-11R-A084-07 BRCA.mRNA 1.841625 0.8096429 3.131333 2.4954167 -1.918500
5 TCGA-A1-A0SK-01A-12R-A084-07 BRCA.mRNA -6.025250 0.2508571 -1.451750 -4.8606667 -1.171500
6 TCGA-A1-A0SM-01A-11R-A084-07 BRCA.mRNA 1.804500 1.3107857 4.041083 2.7970000 3.529750
7 TCGA-A1-A0SO-01A-22R-A084-07 BRCA.mRNA -4.879250 -0.2369286 -0.724750 -4.4860833 -1.455750
8 TCGA-A1-A0SP-01A-11R-A084-07 BRCA.mRNA -3.143250 -1.2432143 -1.193083 -1.6274167 -0.986750
9 TCGA-A2-A04N-01A-11R-A115-07 BRCA.mRNA 2.034000 1.2074286 2.278833 4.1155833 0.668000
10 TCGA-A2-A04P-01A-31R-A034-07 BRCA.mRNA -0.293125 0.2883571 -1.605083 0.4731667 0.011500
# ... with 1,295 more rows To display the number of sample in each data set, type this:
nb_samples <- table(expr$dataset)
nb_samples
BRCA.mRNA LUSC.mRNA OV.mRNA
590 154 561 We can simplify data set names by removing the “mRNA” tag. This can be done using the R base function gsub().
expr$dataset <- gsub(pattern = ".mRNA", replacement = "", expr$dataset)Let’s simplify also the patients’ barcode column. The following R code will change the barcodes into BRCA1, BRCA2, …, OV1, OV2, …., etc
expr$bcr_patient_barcode <- paste0(expr$dataset, c(1:590, 1:561, 1:154))
expr# A tibble: 1,305 x 7
bcr_patient_barcode dataset GATA3 PTEN XBP1 ESR1 MUC1
1 BRCA1 BRCA 2.870500 1.3613571 2.983333 3.0842500 1.652125
2 BRCA2 BRCA 2.166250 0.4283571 2.550833 2.3860000 3.080250
3 BRCA3 BRCA 1.323500 1.3056429 3.020417 0.7912500 2.985250
4 BRCA4 BRCA 1.841625 0.8096429 3.131333 2.4954167 -1.918500
5 BRCA5 BRCA -6.025250 0.2508571 -1.451750 -4.8606667 -1.171500
6 BRCA6 BRCA 1.804500 1.3107857 4.041083 2.7970000 3.529750
7 BRCA7 BRCA -4.879250 -0.2369286 -0.724750 -4.4860833 -1.455750
8 BRCA8 BRCA -3.143250 -1.2432143 -1.193083 -1.6274167 -0.986750
9 BRCA9 BRCA 2.034000 1.2074286 2.278833 4.1155833 0.668000
10 BRCA10 BRCA -0.293125 0.2883571 -1.605083 0.4731667 0.011500
# ... with 1,295 more rows The above (expr) dataset has been saved at https://raw.githubusercontent.com/kassambara/data/master/expr_tcga.txt. This data is required to practice the R code provided in this tutotial.
If you experience some issues in installing the RTCGA packages, You can simply load the data as follow:
expr <- read.delim("https://raw.githubusercontent.com/kassambara/data/master/expr_tcga.txt",
stringsAsFactors = FALSE)Box plots
(ggplot2 way of creating box plot)
Create a box plot of a gene expression profile, colored by groups (here data set/cancer type):
library(ggpubr)
# GATA3
ggboxplot(expr, x = "dataset", y = "GATA3",
title = "GATA3", ylab = "Expression",
color = "dataset", palette = "jco")
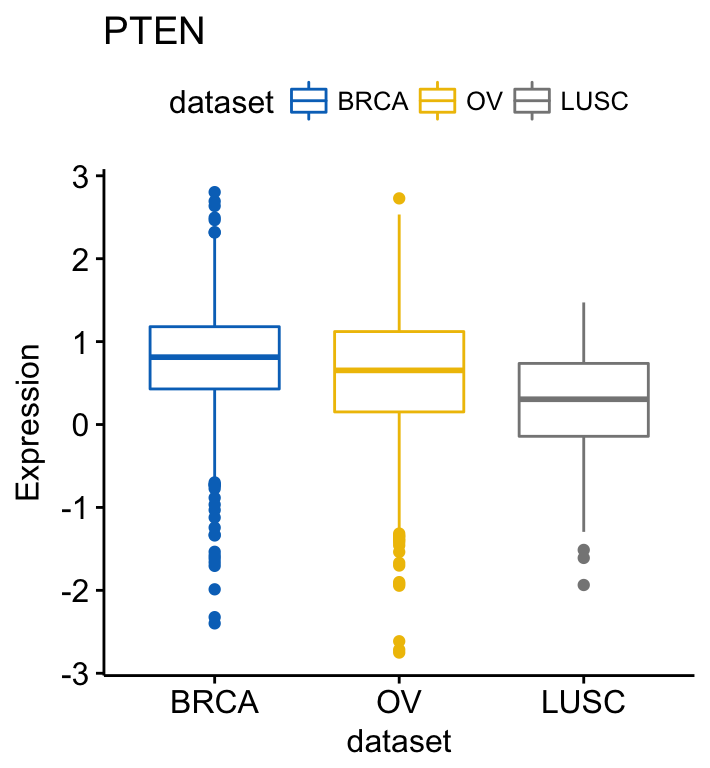
# PTEN
ggboxplot(expr, x = "dataset", y = "PTEN",
title = "PTEN", ylab = "Expression",
color = "dataset", palette = "jco")Clik here to view.
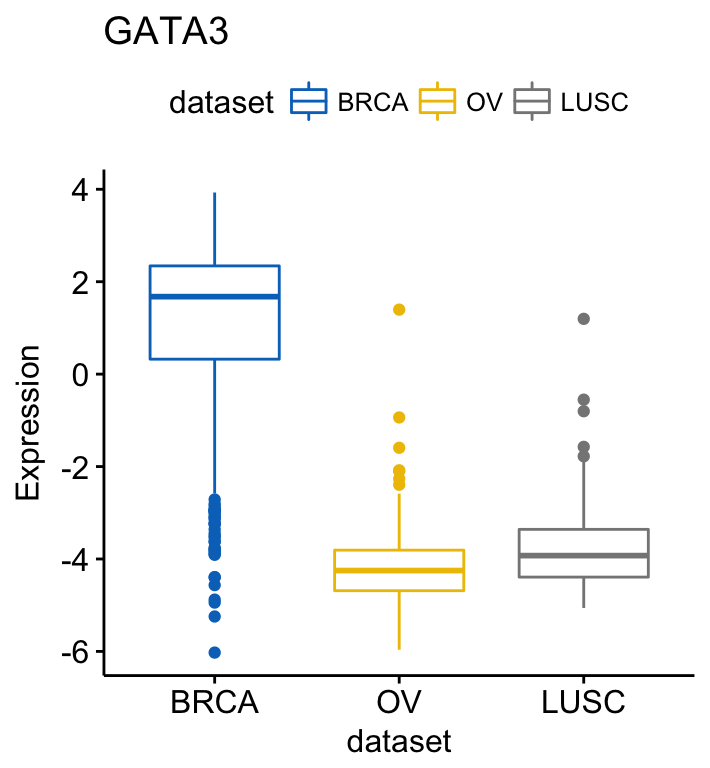 Image may be NSFW.
Image may be NSFW.Clik here to view.
Exploratory Data visualization: Gene Expression Data
Note that, the argument palette is used to change color palettes. Allowed values include:
- “grey” for grey color palettes;
- brewer palettes e.g. “RdBu”, “Blues”, …;. To view all, type this in R: RColorBrewer::display.brewer.all() or click here to see all brewer palettes;
- or custom color palettes e.g. c(“blue”, “red”) or c(“#00AFBB”, “#E7B800”);
- and scientific journal palettes from the ggsci R package, e.g.: “npg”, “aaas”, “lancet”, “jco”, “ucscgb”, “uchicago”, “simpsons” and “rickandmorty”.
Instead of repeating the same R code for each gene, you can create a list of plots at once, as follow:
# Create a list of plots
p <- ggboxplot(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
title = c("GATA3", "PTEN", "XBP1"),
ylab = "Expression",
color = "dataset", palette = "jco")
# View GATA3
p$GATA3
# View PTEN
p$PTEN
# View XBP1
p$XBP1Note that, when the argument y contains multiple variables (here multiple gene names), then the arguments title, xlab and ylab can be also a character vector of same length as y.
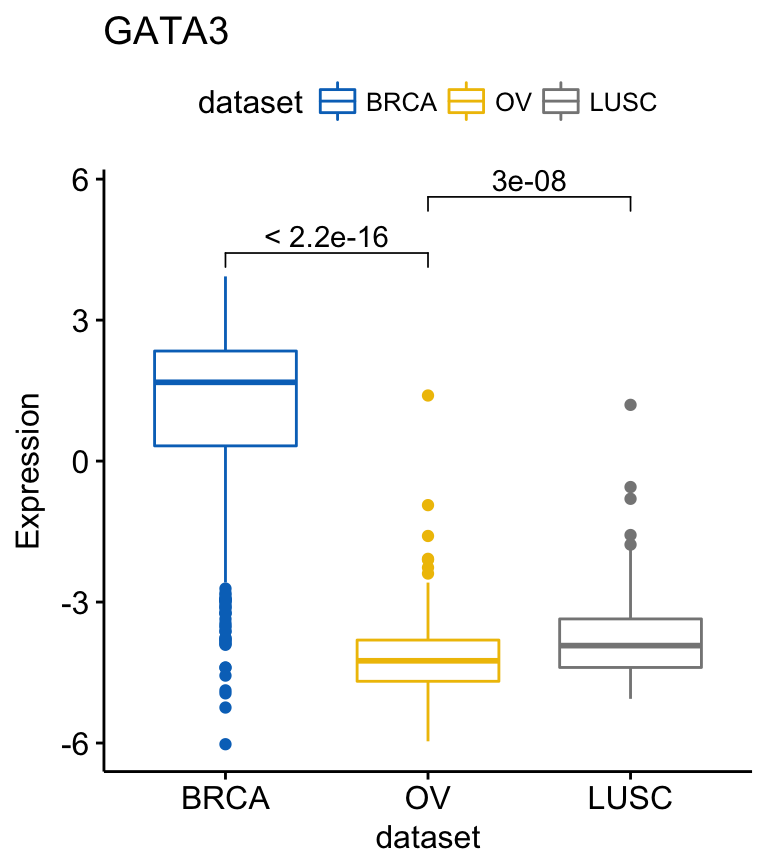
To add p-values and significance levels to the boxplots, read our previous article: Add P-values and Significance Levels to ggplots. Briefly, you can do this:
my_comparisons <- list(c("BRCA", "OV"), c("OV", "LUSC"))
ggboxplot(expr, x = "dataset", y = "GATA3",
title = "GATA3", ylab = "Expression",
color = "dataset", palette = "jco")+
stat_compare_means(comparisons = my_comparisons)Clik here to view.
Exploratory Data visualization: Gene Expression Data
For each of the genes, you can compare the different groups as follow:
compare_means(c(GATA3, PTEN, XBP1) ~ dataset, data = expr)# A tibble: 9 x 8
.y. group1 group2 p p.adj p.format p.signif method
1 GATA3 BRCA OV 1.111768e-177 3.335304e-177 < 2e-16 **** Wilcoxon
2 GATA3 BRCA LUSC 6.684016e-73 1.336803e-72 < 2e-16 **** Wilcoxon
3 GATA3 OV LUSC 2.965702e-08 2.965702e-08 3.0e-08 **** Wilcoxon
4 PTEN BRCA OV 6.791940e-05 6.791940e-05 6.8e-05 **** Wilcoxon
5 PTEN BRCA LUSC 1.042830e-16 3.128489e-16 < 2e-16 **** Wilcoxon
6 PTEN OV LUSC 1.280576e-07 2.561153e-07 1.3e-07 **** Wilcoxon
7 XBP1 BRCA OV 2.551228e-123 7.653685e-123 < 2e-16 **** Wilcoxon
8 XBP1 BRCA LUSC 1.950162e-42 3.900324e-42 < 2e-16 **** Wilcoxon
9 XBP1 OV LUSC 4.239570e-11 4.239570e-11 4.2e-11 **** Wilcoxon If you want to select items (here cancer types) to display or to remove a particular item from the plot, use the argument select or remove, as follow:
# Select BRCA and OV cancer types
ggboxplot(expr, x = "dataset", y = "GATA3",
title = "GATA3", ylab = "Expression",
color = "dataset", palette = "jco",
select = c("BRCA", "OV"))
# or remove BRCA
ggboxplot(expr, x = "dataset", y = "GATA3",
title = "GATA3", ylab = "Expression",
color = "dataset", palette = "jco",
remove = "BRCA")Clik here to view.
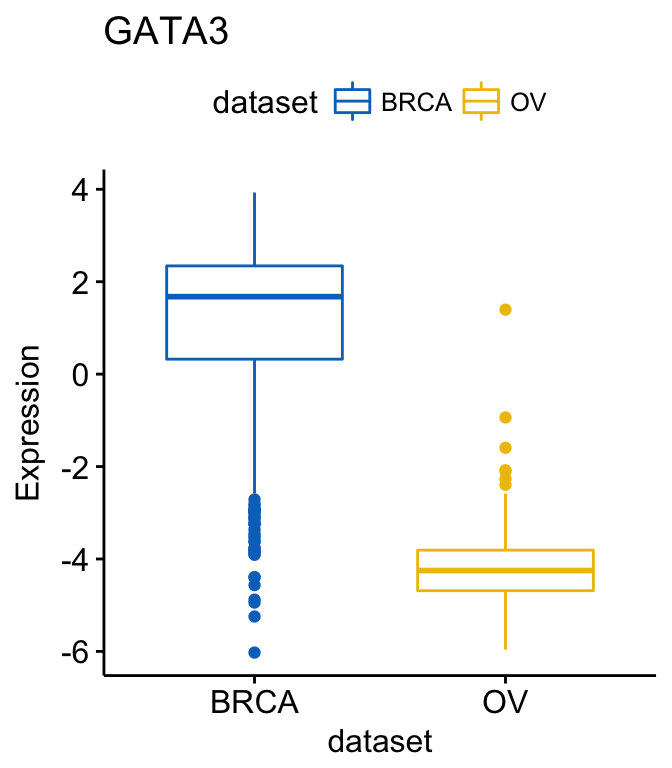 Image may be NSFW.
Image may be NSFW.Clik here to view.
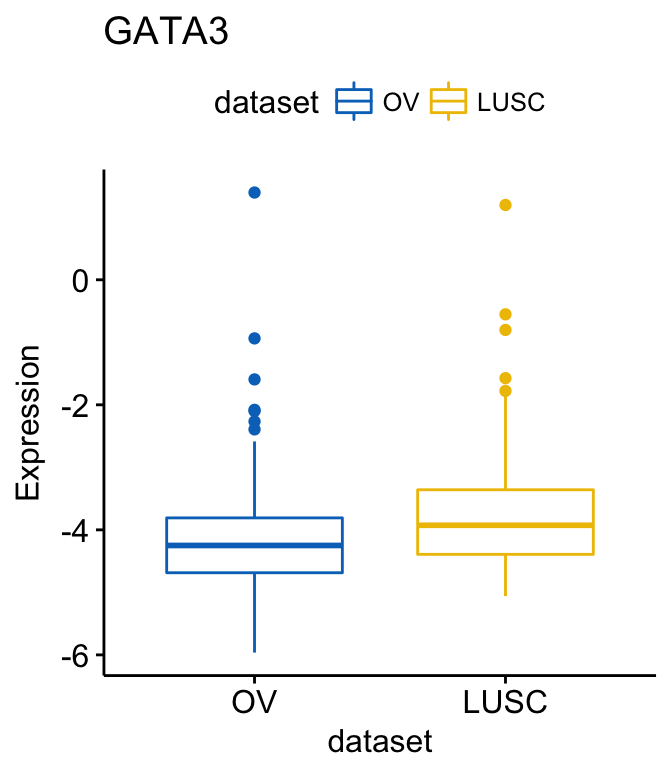
Exploratory Data visualization: Gene Expression Data
To change the order of the data sets on x axis, use the argument order. For example order = c(“LUSC”, “OV”, “BRCA”):
# Order data sets
ggboxplot(expr, x = "dataset", y = "GATA3",
title = "GATA3", ylab = "Expression",
color = "dataset", palette = "jco",
order = c("LUSC", "OV", "BRCA"))Clik here to view.
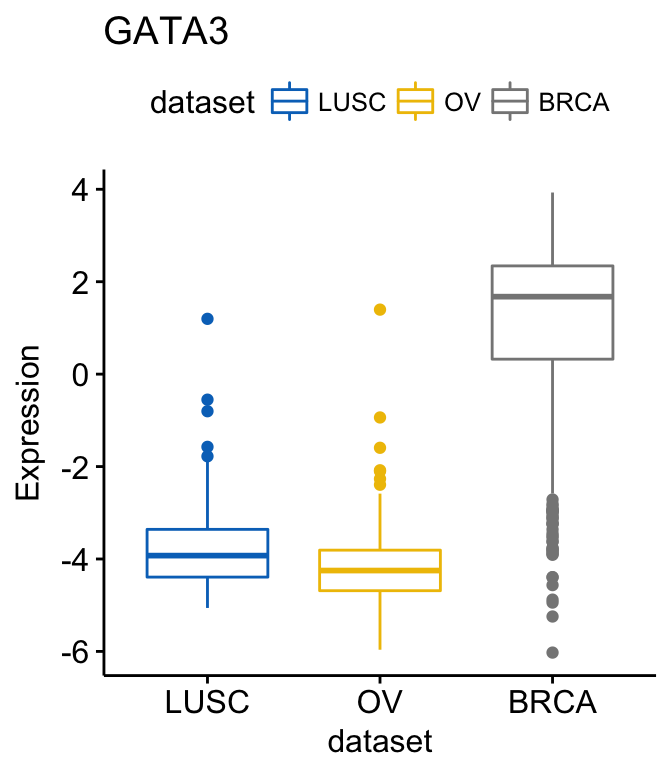
Exploratory Data visualization: Gene Expression Data
To create horizontal plots, use the argument rotate = TRUE:
ggboxplot(expr, x = "dataset", y = "GATA3",
title = "GATA3", ylab = "Expression",
color = "dataset", palette = "jco",
rotate = TRUE)Clik here to view.
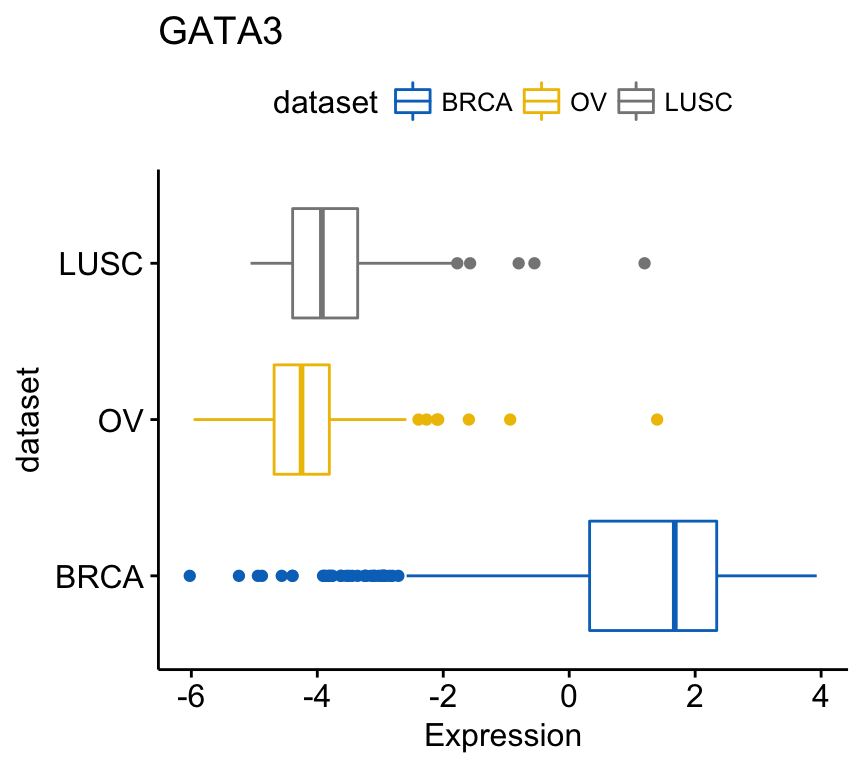
Exploratory Data visualization: Gene Expression Data
To combine the three gene expression plots into a multi-panel plot, use the argument combine = TRUE:
ggboxplot(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
combine = TRUE,
ylab = "Expression",
color = "dataset", palette = "jco")Clik here to view.
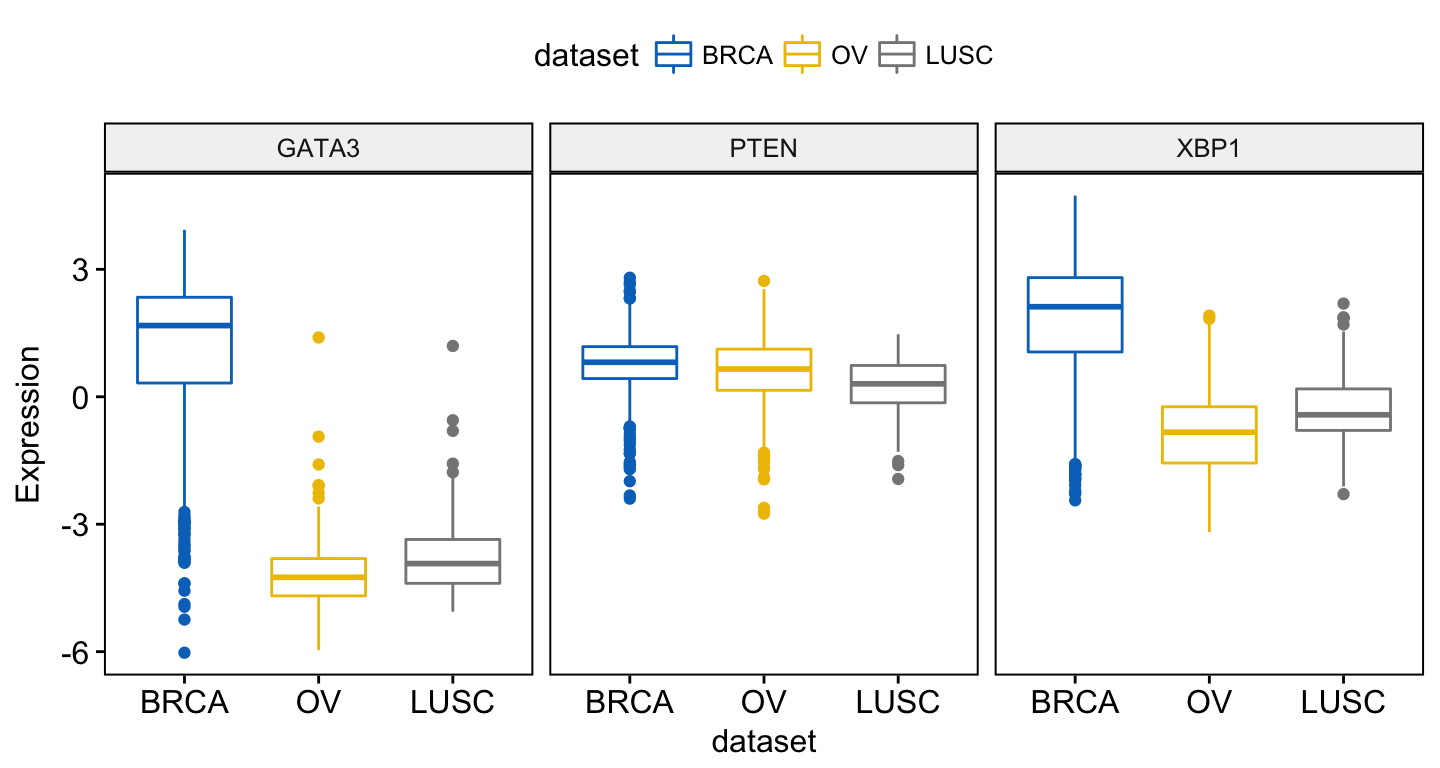
Exploratory Data visualization: Gene Expression Data
You can also merge the 3 plots using the argument merge = TRUE or merge = “asis”:
ggboxplot(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
merge = TRUE,
ylab = "Expression",
palette = "jco")Clik here to view.
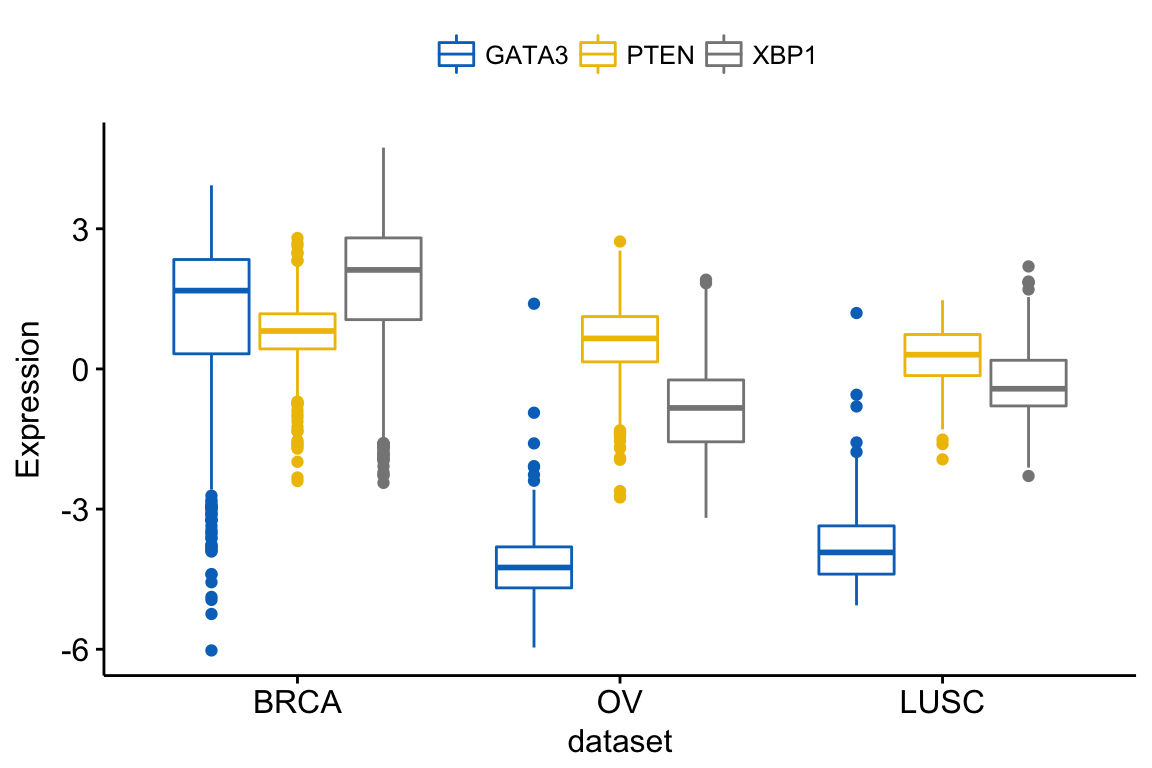
Exploratory Data visualization: Gene Expression Data
In the plot above, It’s easy to visually compare the expression level of the different genes in each cancer type.
But you might want to put genes (y variables) on x axis, in order to compare the expression level in the different cell subpopulations.
In this situation, the y variables (i.e.: genes) become x tick labels and the x variable (i.e.: dataset) becomes the grouping variable. To do this, use the argument merge = “flip”.
ggboxplot(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
merge = "flip",
ylab = "Expression",
palette = "jco")Clik here to view.
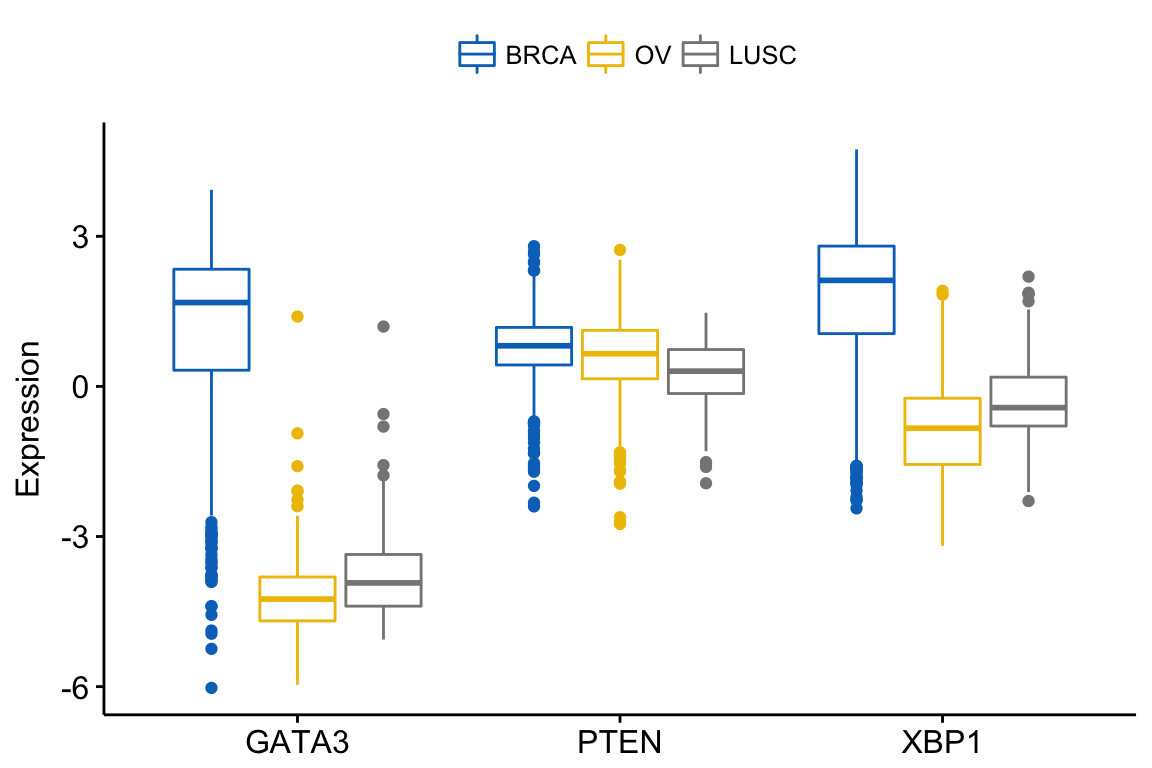
Exploratory Data visualization: Gene Expression Data
You might want to add jittered points on the boxplot. Each point correspond to individual observations. To add jittered points, use the argument add = “jitter” as follow. To customize the added elements, specify the argument add.params.
ggboxplot(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
combine = TRUE,
color = "dataset", palette = "jco",
ylab = "Expression",
add = "jitter", # Add jittered points
add.params = list(size = 0.1, jitter = 0.2) # Point size and the amount of jittering
)Clik here to view.
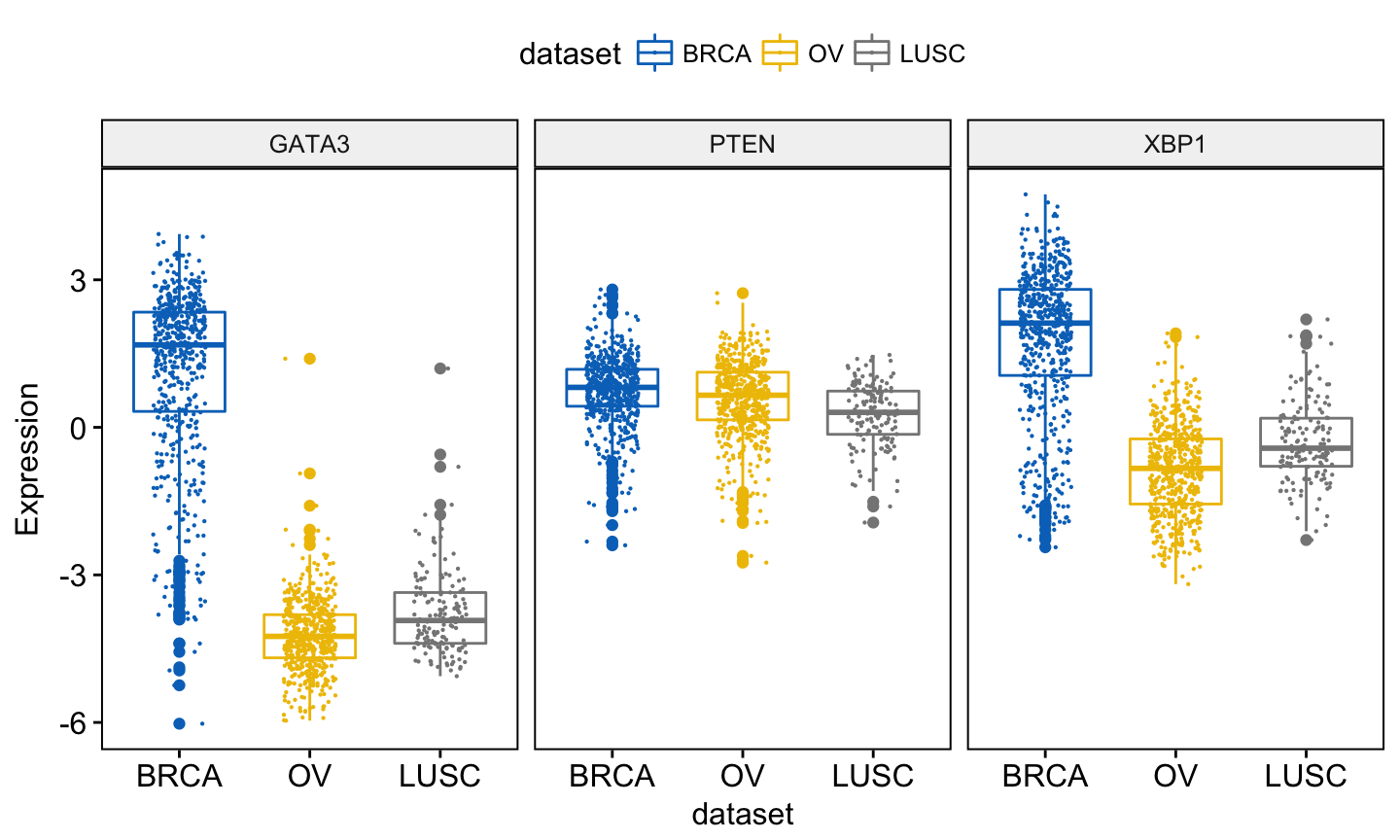
Exploratory Data visualization: Gene Expression Data
Note that, when using ggboxplot() sensible values for the argument add are one of c(“jitter”, “dotplot”). If you decide to use add = “dotplot”, you can adjust dotsize and binwidth wen you have a strong dense dotplot. Read more about binwidth.
You can add and adjust a dotplot as follow:
ggboxplot(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
combine = TRUE,
color = "dataset", palette = "jco",
ylab = "Expression",
add = "dotplot", # Add dotplot
add.params = list(binwidth = 0.1, dotsize = 0.3)
)Clik here to view.
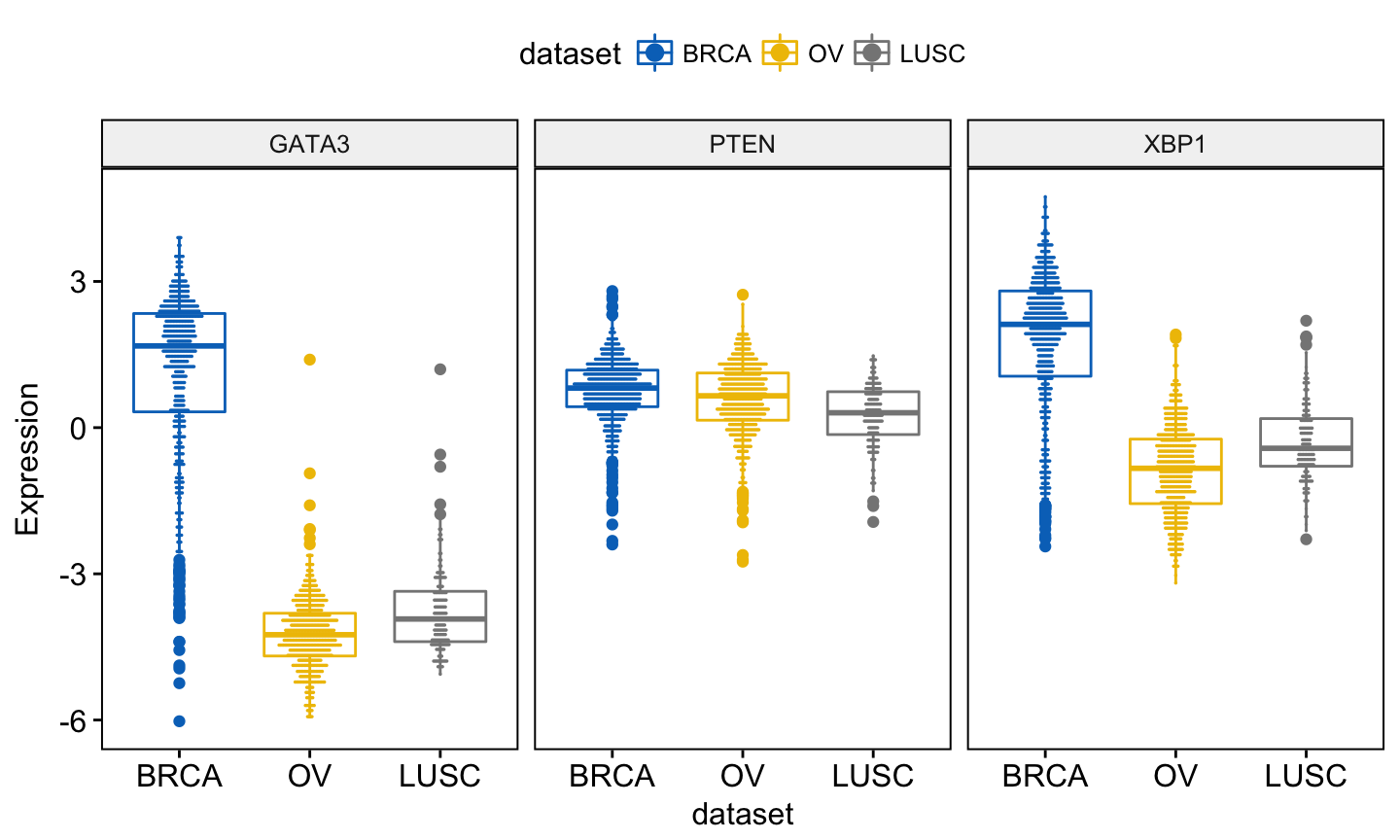
Exploratory Data visualization: Gene Expression Data
You might want to label the boxplot by showing the names of samples with the top n highest or lowest values. In this case, you can use the following arguments:
- label: the name of the column containing point labels.
- label.select: can be of two formats:
- a character vector specifying some labels to show.
- a list containing one or the combination of the following components:
- top.up and top.down: to display the labels of the top up/down points. For example, label.select = list(top.up = 10, top.down = 4).
- criteria: to filter, for example, by x and y variables values, use this: label.select = list(criteria = “`y` > 3.9 & `y` < 5 & `x` %in% c(‘BRCA’, ‘OV’)”).
For example:
ggboxplot(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
combine = TRUE,
color = "dataset", palette = "jco",
ylab = "Expression",
add = "jitter", # Add jittered points
add.params = list(size = 0.1, jitter = 0.2), # Point size and the amount of jittering
label = "bcr_patient_barcode", # column containing point labels
label.select = list(top.up = 2, top.down = 2),# Select some labels to display
font.label = list(size = 9, face = "italic"), # label font
repel = TRUE # Avoid label text overplotting
)Clik here to view.
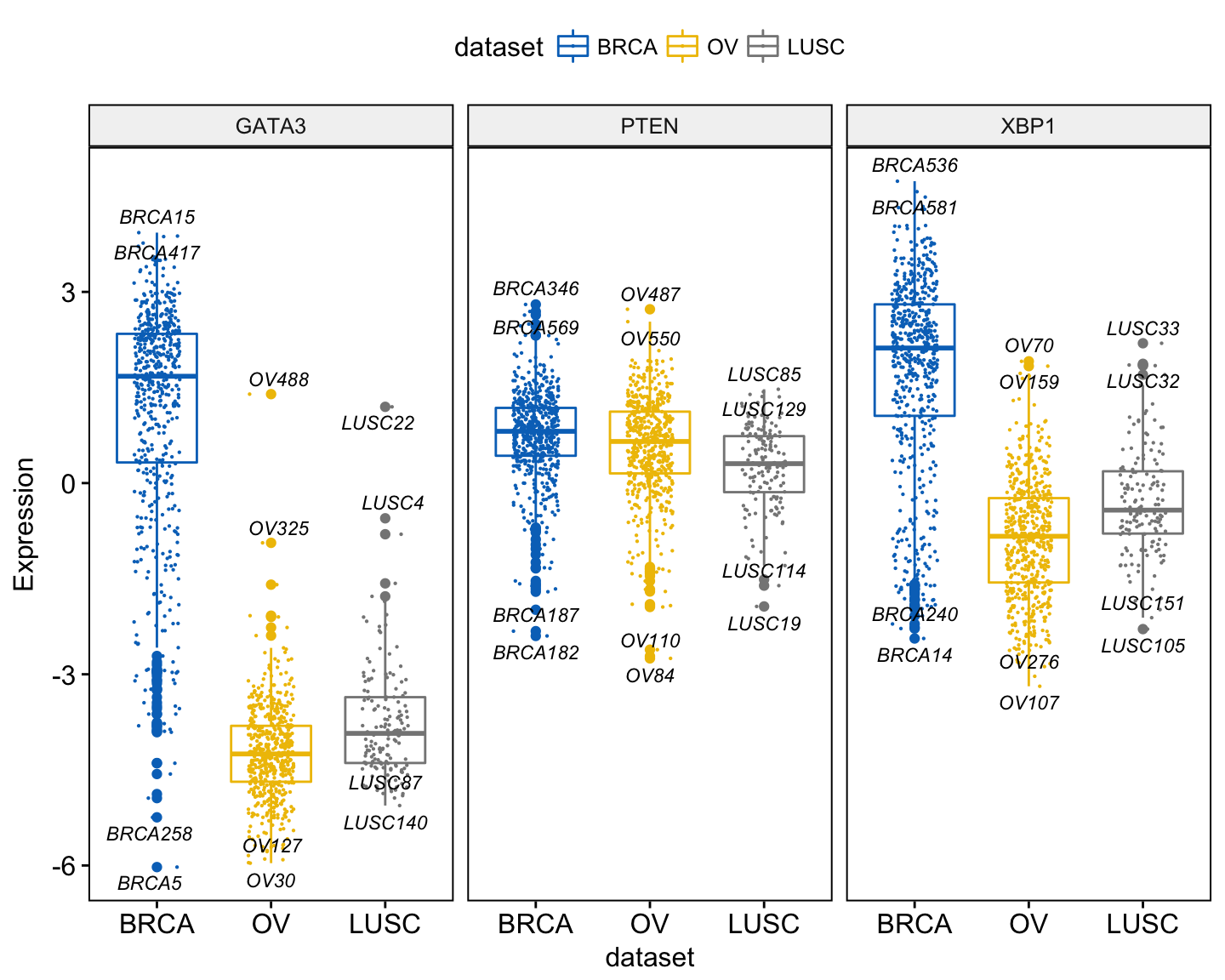
Exploratory Data visualization: Gene Expression Data
A complex criteria for labeling can be specified as follow:
label.select.criteria <- list(criteria = "`y` > 3.9 & `x` %in% c('BRCA', 'OV')")
ggboxplot(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
combine = TRUE,
color = "dataset", palette = "jco",
ylab = "Expression",
label = "bcr_patient_barcode", # column containing point labels
label.select = label.select.criteria, # Select some labels to display
font.label = list(size = 9, face = "italic"), # label font
repel = TRUE # Avoid label text overplotting
)Other types of plots, with the same arguments as the function ggboxplot(), are available, such as stripchart and violin plots.
Violin plots
(ggplot2 way of creating violin plot)
The following R code draws violin plots with box plots inside:
ggviolin(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
combine = TRUE,
color = "dataset", palette = "jco",
ylab = "Expression",
add = "boxplot")Clik here to view.
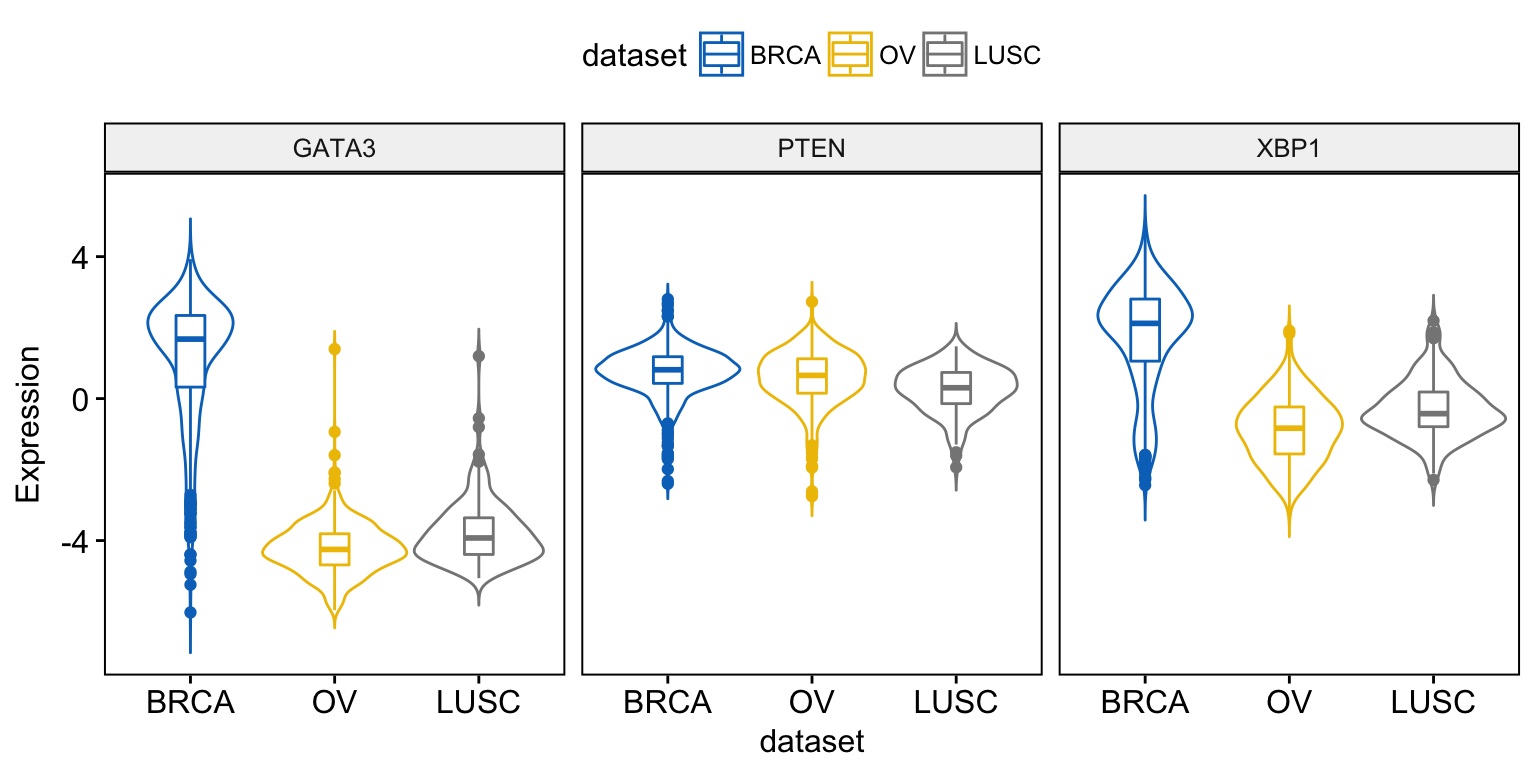
Exploratory Data visualization: Gene Expression Data
Instead of adding a box plot inside the violin plot, you can add the median + interquantile range as follow:
ggviolin(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
combine = TRUE,
color = "dataset", palette = "jco",
ylab = "Expression",
add = "median_iqr")Clik here to view.
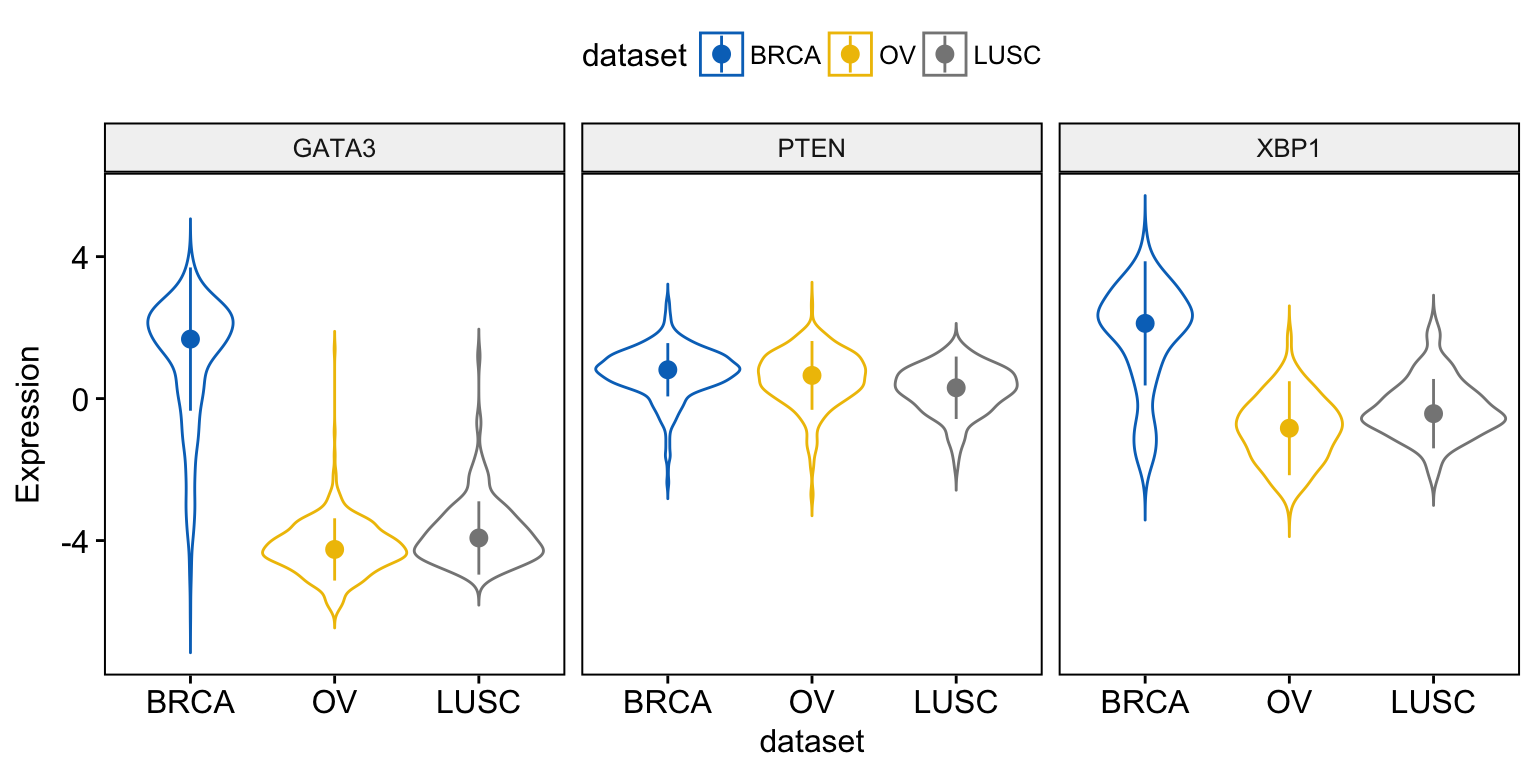
Exploratory Data visualization: Gene Expression Data
When using the function ggviolin(), sensible values for the argument add include: “mean”, “mean_se”, “mean_sd”, “mean_ci”, “mean_range”, “median”, “median_iqr”, “median_mad”, “median_range”.
You can also add “jitter” points and “dotplot” inside the violin plot as described previously in the box plot section.
Stripcharts and dot plots
To draw a stripchart, type this:
ggstripchart(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
combine = TRUE,
color = "dataset", palette = "jco",
size = 0.1, jitter = 0.2,
ylab = "Expression",
add = "median_iqr",
add.params = list(color = "gray"))Clik here to view.
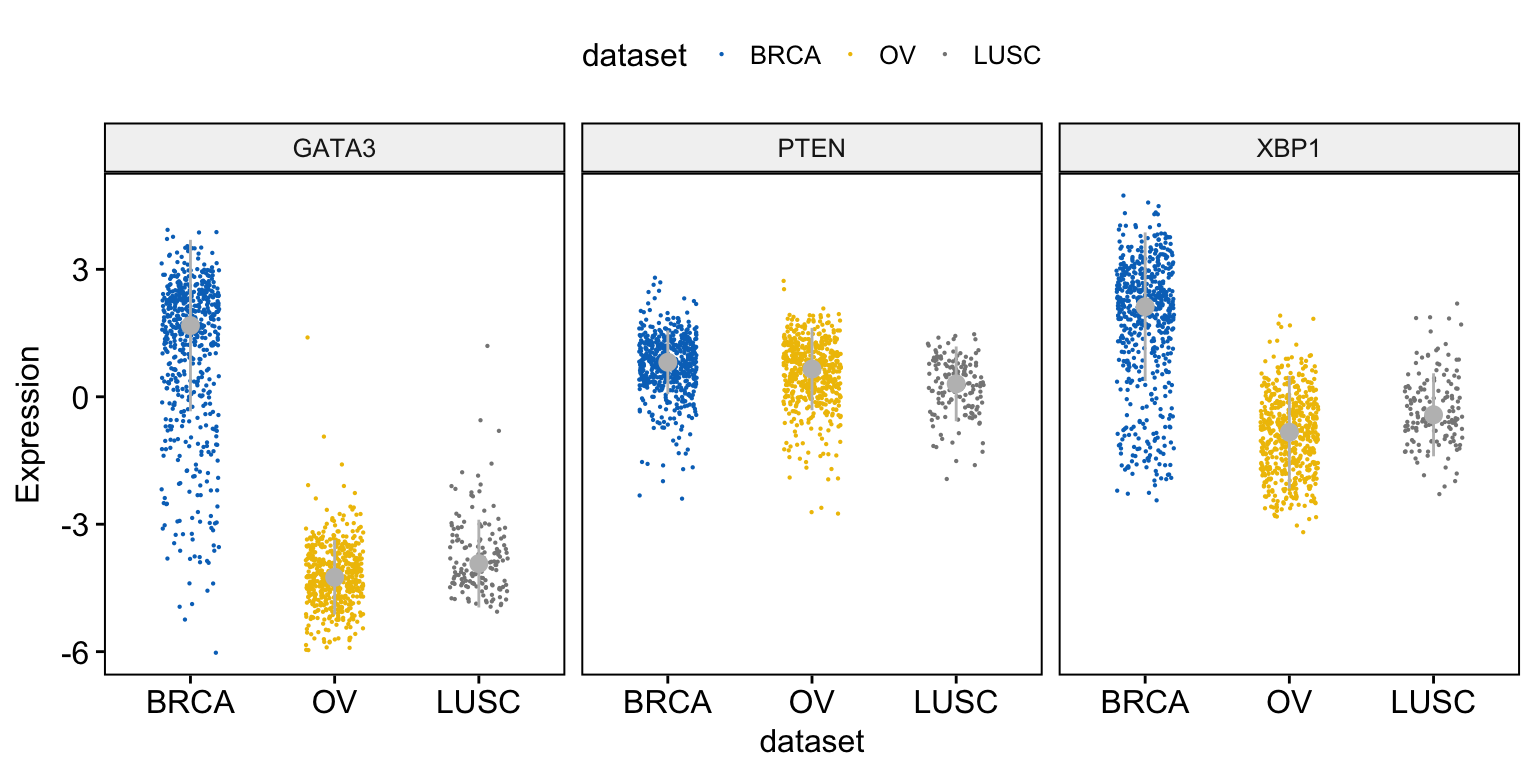
Exploratory Data visualization: Gene Expression Data
(ggplot2 way of creating stripcharts)
For a dot plot, use this:
ggdotplot(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
combine = TRUE,
color = "dataset", palette = "jco",
fill = "white",
binwidth = 0.1,
ylab = "Expression",
add = "median_iqr",
add.params = list(size = 0.9))Clik here to view.
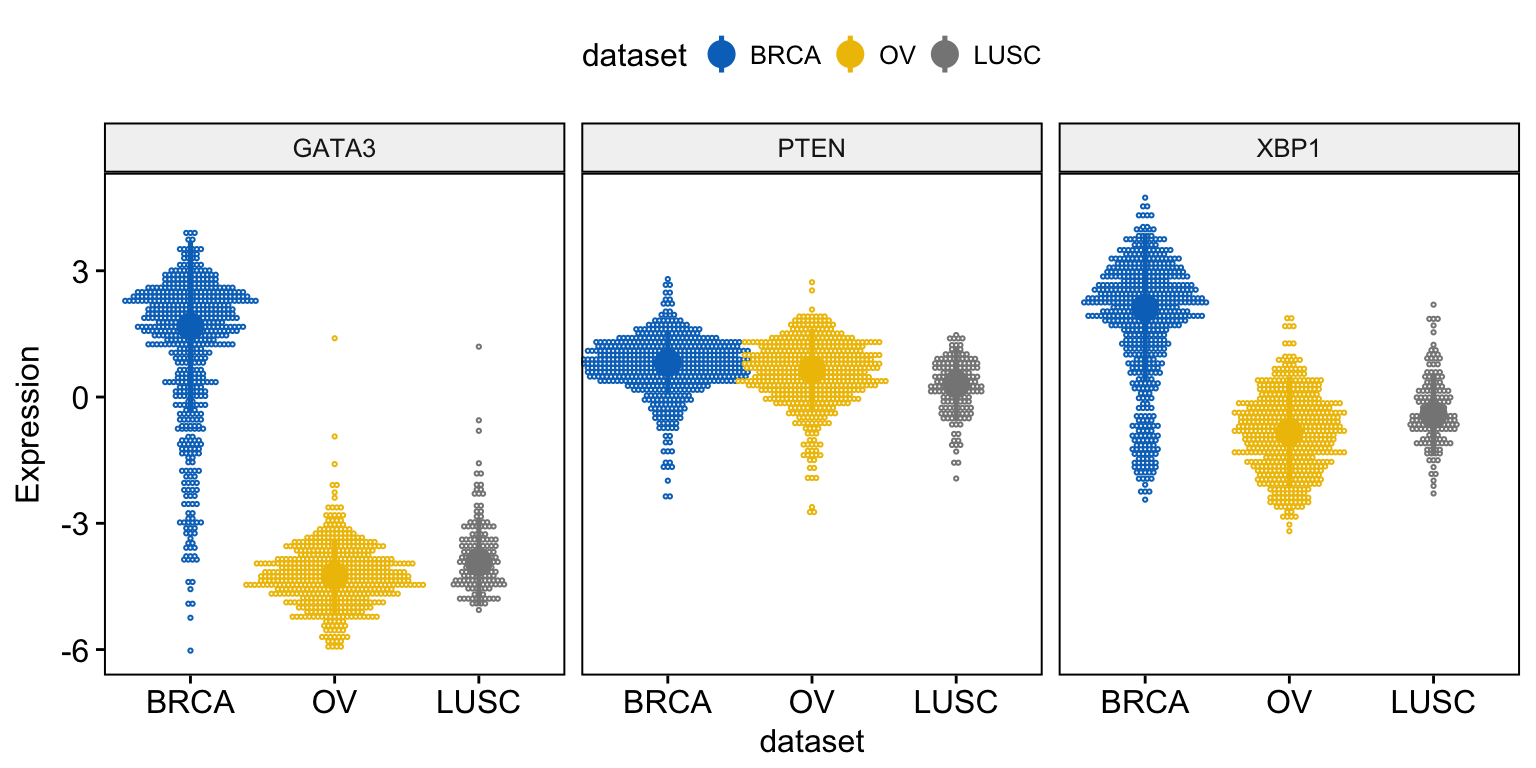
Exploratory Data visualization: Gene Expression Data
Density plots
(ggplot2 way of creating density plots)
To visualize the distribution as a density plot, use the function ggdensity() as follow:
# Basic density plot
ggdensity(expr,
x = c("GATA3", "PTEN", "XBP1"),
y = "..density..",
combine = TRUE, # Combine the 3 plots
xlab = "Expression",
add = "median", # Add median line.
rug = TRUE # Add marginal rug
)Clik here to view.
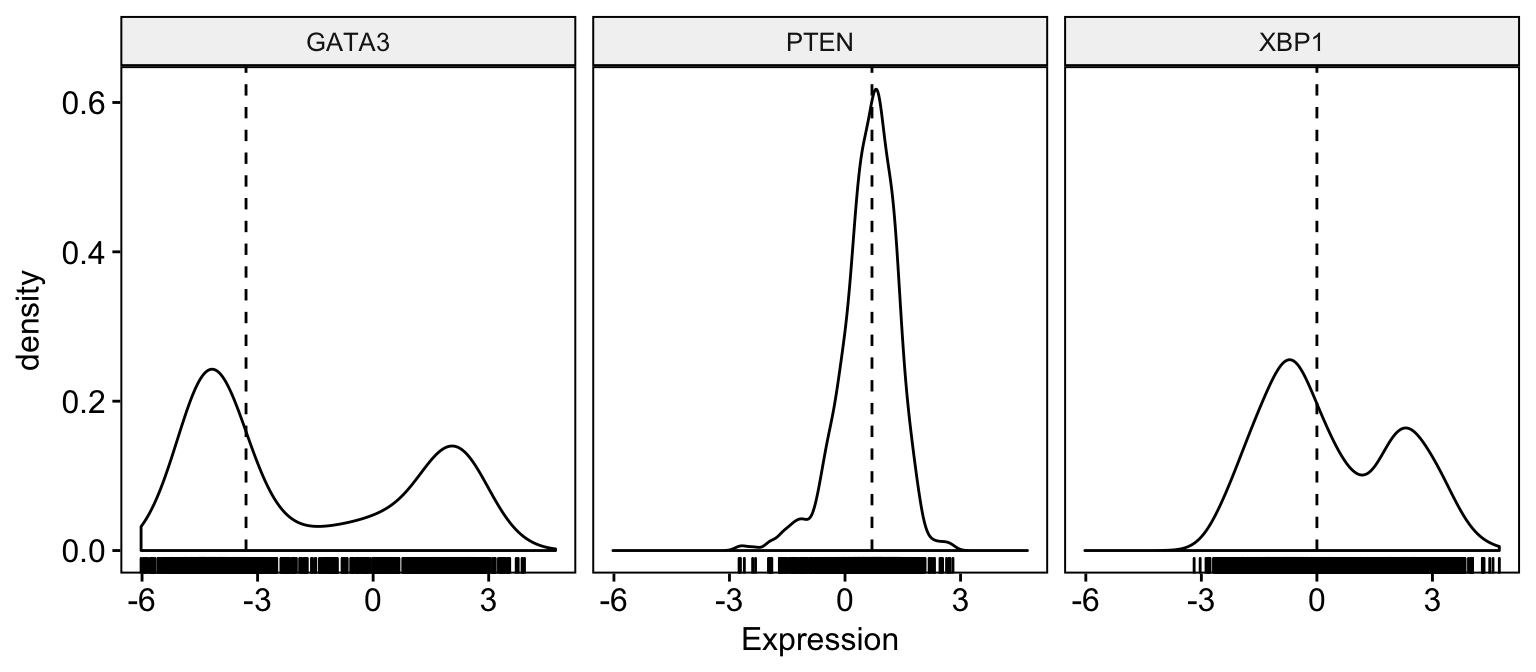
Exploratory Data visualization: Gene Expression Data
# Change color and fill by dataset
ggdensity(expr,
x = c("GATA3", "PTEN", "XBP1"),
y = "..density..",
combine = TRUE, # Combine the 3 plots
xlab = "Expression",
add = "median", # Add median line.
rug = TRUE, # Add marginal rug
color = "dataset",
fill = "dataset",
palette = "jco"
)Clik here to view.
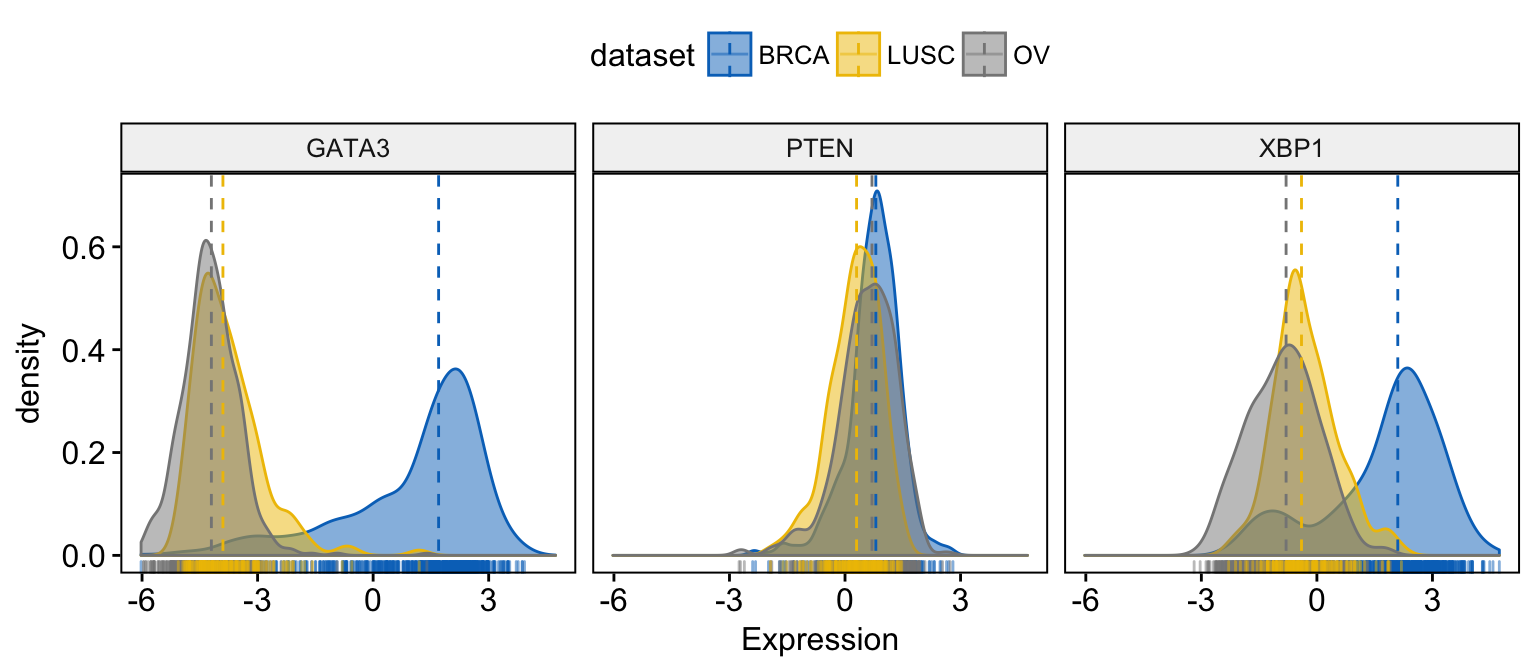
Exploratory Data visualization: Gene Expression Data
# Merge the 3 plots
# and use y = "..count.." instead of "..density.."
ggdensity(expr,
x = c("GATA3", "PTEN", "XBP1"),
y = "..count..",
merge = TRUE, # Merge the 3 plots
xlab = "Expression",
add = "median", # Add median line.
rug = TRUE , # Add marginal rug
palette = "jco" # Change color palette
)Clik here to view.
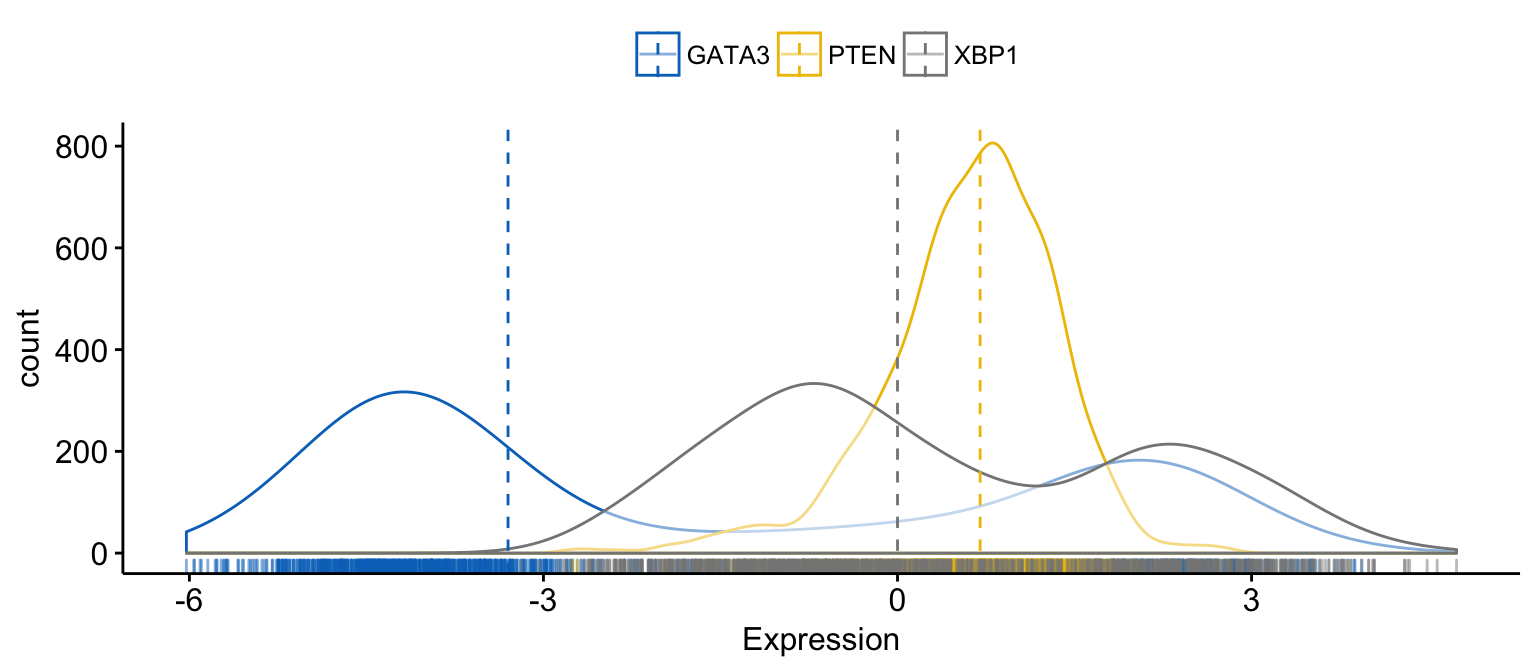
Exploratory Data visualization: Gene Expression Data
# color and fill by x variables
ggdensity(expr,
x = c("GATA3", "PTEN", "XBP1"),
y = "..count..",
color = ".x.", fill = ".x.", # color and fill by x variables
merge = TRUE, # Merge the 3 plots
xlab = "Expression",
add = "median", # Add median line.
rug = TRUE , # Add marginal rug
palette = "jco" # Change color palette
)Clik here to view.
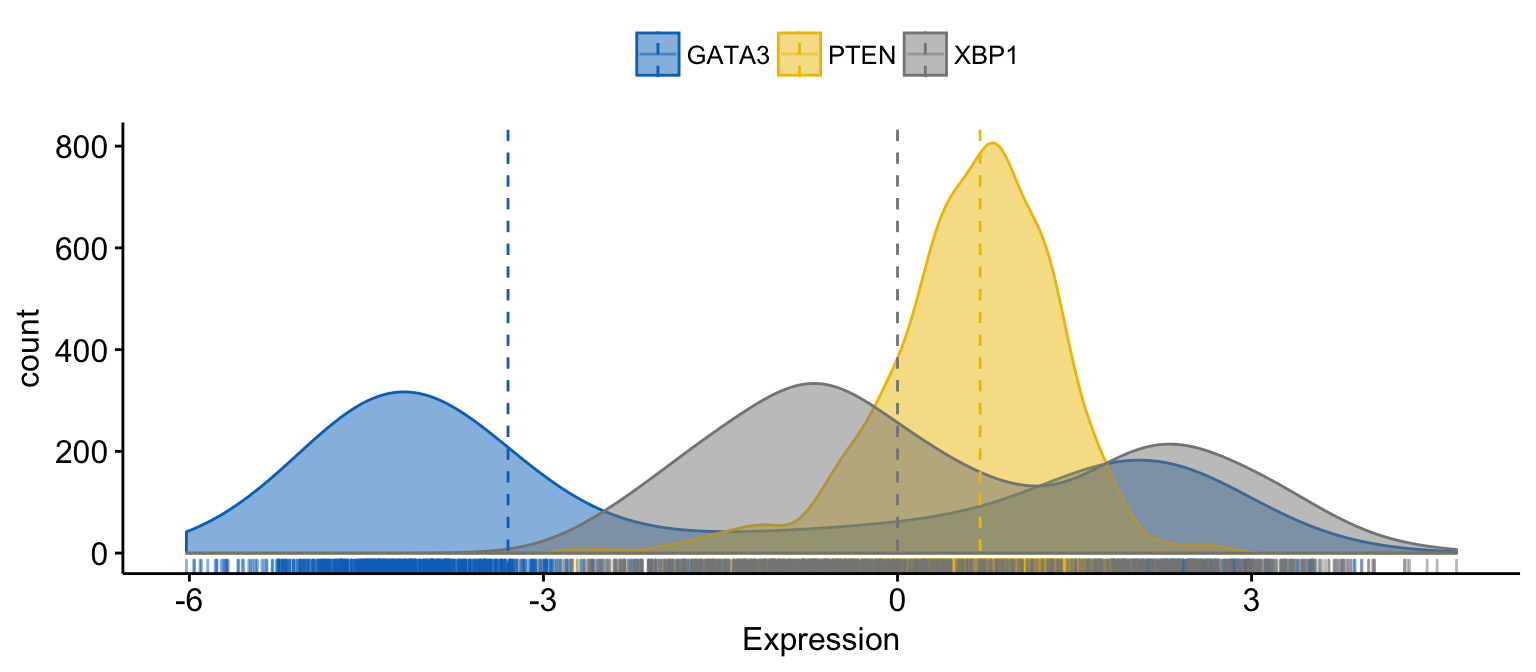
Exploratory Data visualization: Gene Expression Data
# Facet by "dataset"
ggdensity(expr,
x = c("GATA3", "PTEN", "XBP1"),
y = "..count..",
color = ".x.", fill = ".x.",
facet.by = "dataset", # Split by "dataset" into multi-panel
merge = TRUE, # Merge the 3 plots
xlab = "Expression",
add = "median", # Add median line.
rug = TRUE , # Add marginal rug
palette = "jco" # Change color palette
)Clik here to view.
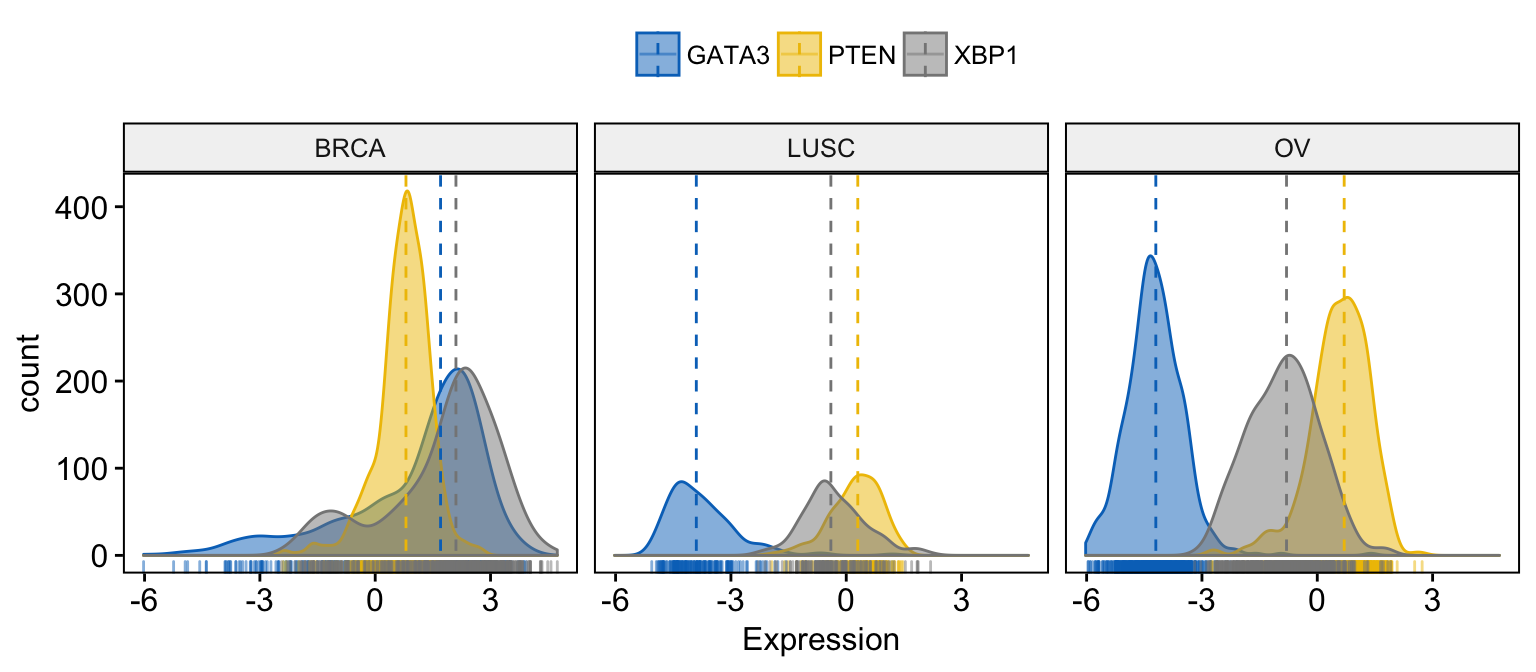
Exploratory Data visualization: Gene Expression Data
Histogram plots
(ggplot2 way of creating histogram plots)
To visualize the distribution as a histogram plot, use the function gghistogram() as follow:
# Basic histogram plot
gghistogram(expr,
x = c("GATA3", "PTEN", "XBP1"),
y = "..density..",
combine = TRUE, # Combine the 3 plots
xlab = "Expression",
add = "median", # Add median line.
rug = TRUE # Add marginal rug
)Clik here to view.
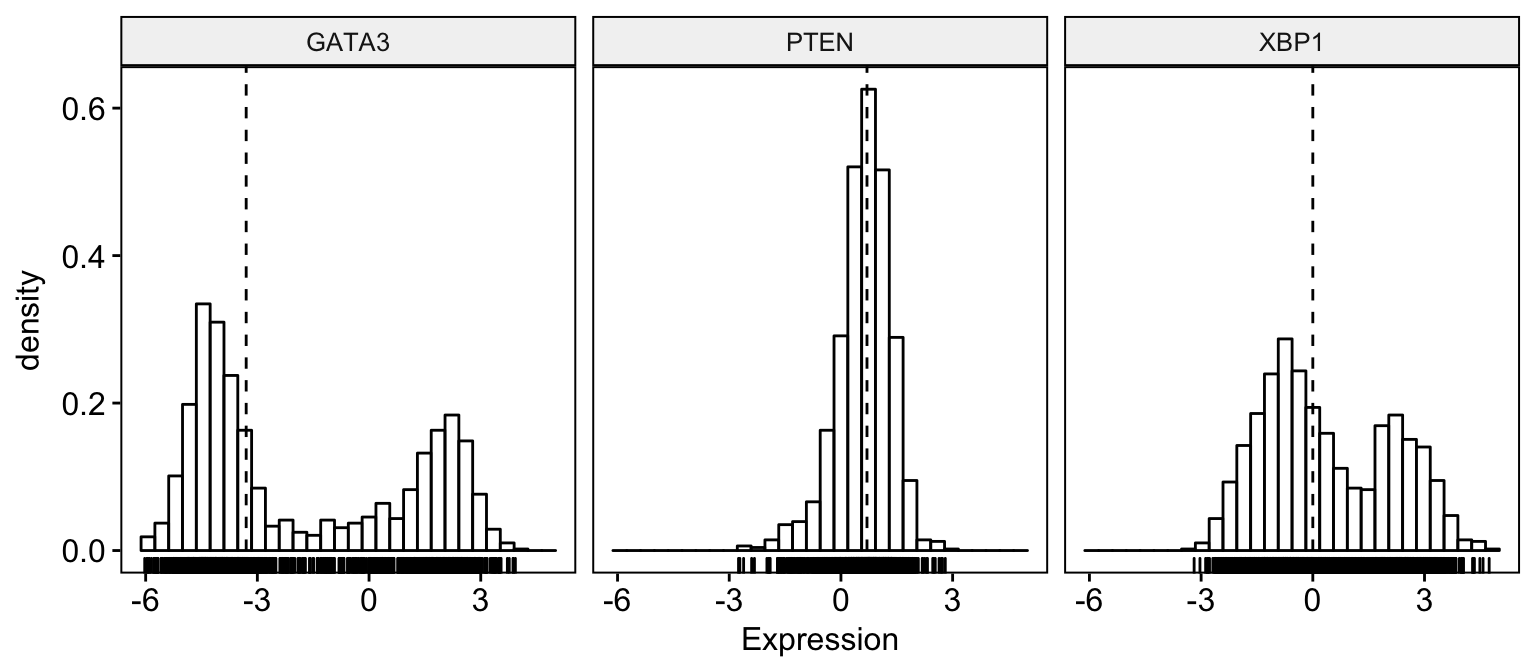
Exploratory Data visualization: Gene Expression Data
# Change color and fill by dataset
gghistogram(expr,
x = c("GATA3", "PTEN", "XBP1"),
y = "..density..",
combine = TRUE, # Combine the 3 plots
xlab = "Expression",
add = "median", # Add median line.
rug = TRUE, # Add marginal rug
color = "dataset",
fill = "dataset",
palette = "jco"
)Clik here to view.
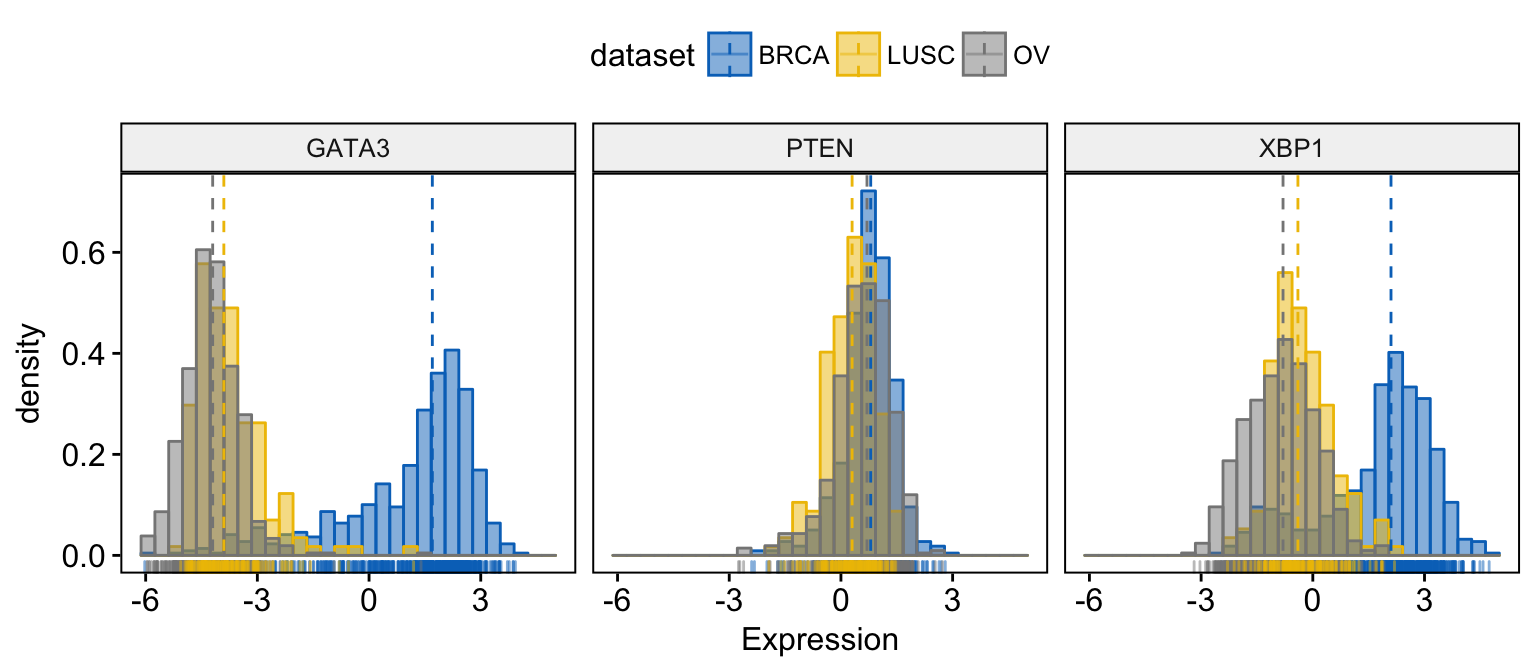
Exploratory Data visualization: Gene Expression Data
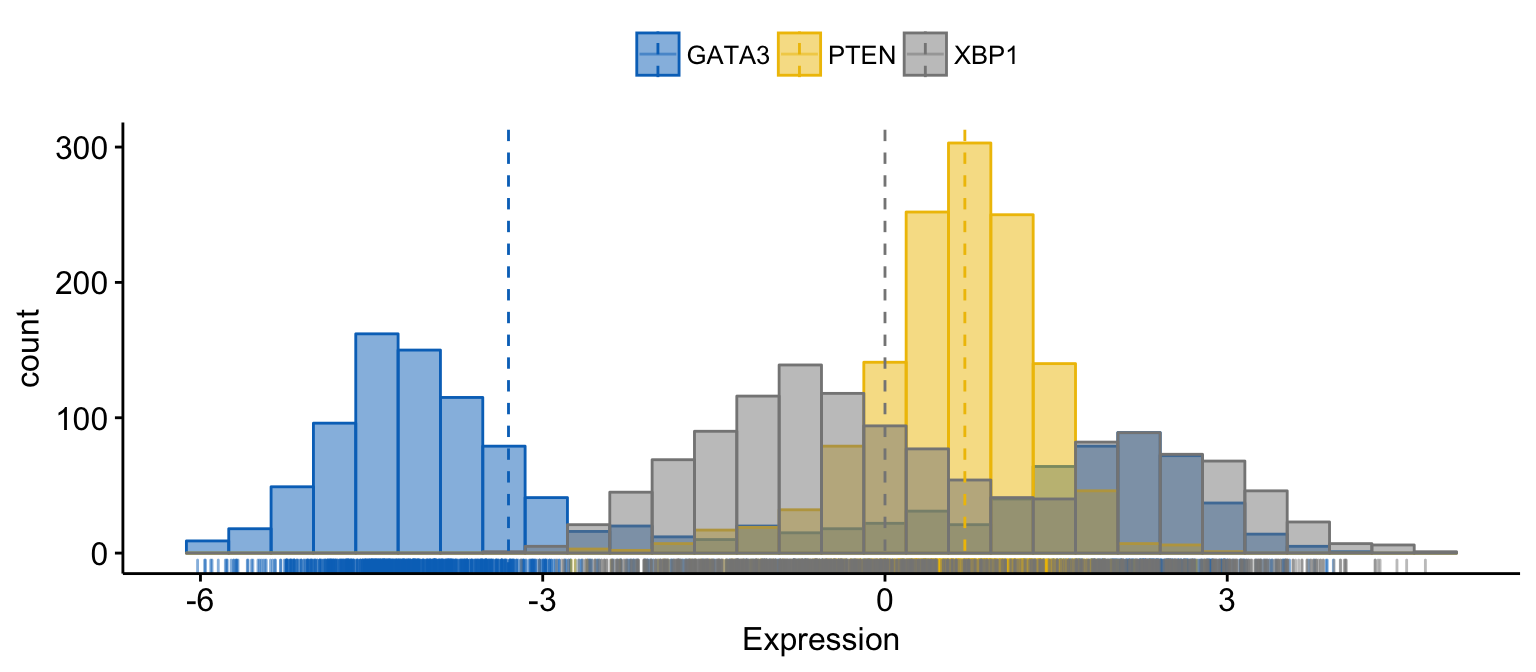
# Merge the 3 plots
# and use y = "..count.." instead of "..density.."
gghistogram(expr,
x = c("GATA3", "PTEN", "XBP1"),
y = "..count..",
merge = TRUE, # Merge the 3 plots
xlab = "Expression",
add = "median", # Add median line.
rug = TRUE , # Add marginal rug
palette = "jco" # Change color palette
)Clik here to view.
Exploratory Data visualization: Gene Expression Data
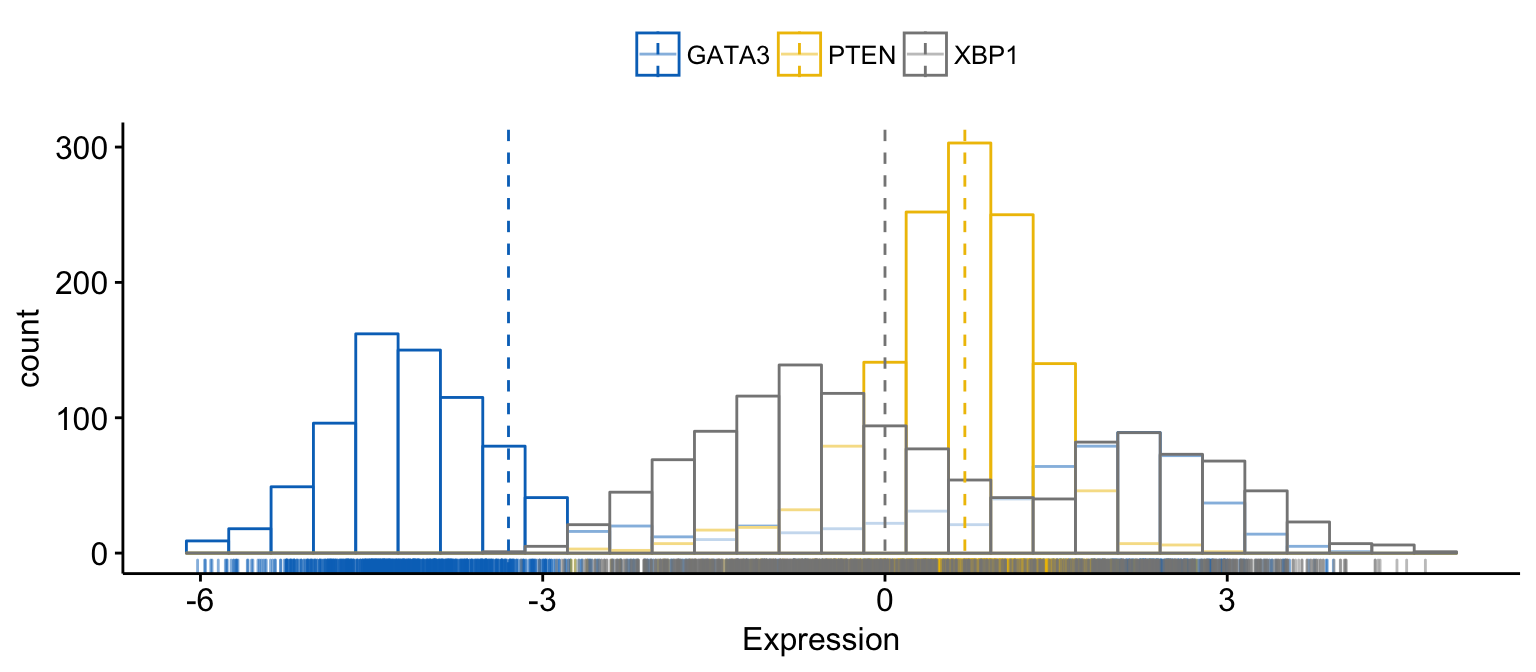
# color and fill by x variables
gghistogram(expr,
x = c("GATA3", "PTEN", "XBP1"),
y = "..count..",
color = ".x.", fill = ".x.", # color and fill by x variables
merge = TRUE, # Merge the 3 plots
xlab = "Expression",
add = "median", # Add median line.
rug = TRUE , # Add marginal rug
palette = "jco" # Change color palette
)Clik here to view.
Exploratory Data visualization: Gene Expression Data
# Facet by "dataset"
gghistogram(expr,
x = c("GATA3", "PTEN", "XBP1"),
y = "..count..",
color = ".x.", fill = ".x.",
facet.by = "dataset", # Split by "dataset" into multi-panel
merge = TRUE, # Merge the 3 plots
xlab = "Expression",
add = "median", # Add median line.
rug = TRUE , # Add marginal rug
palette = "jco" # Change color palette
)Clik here to view.
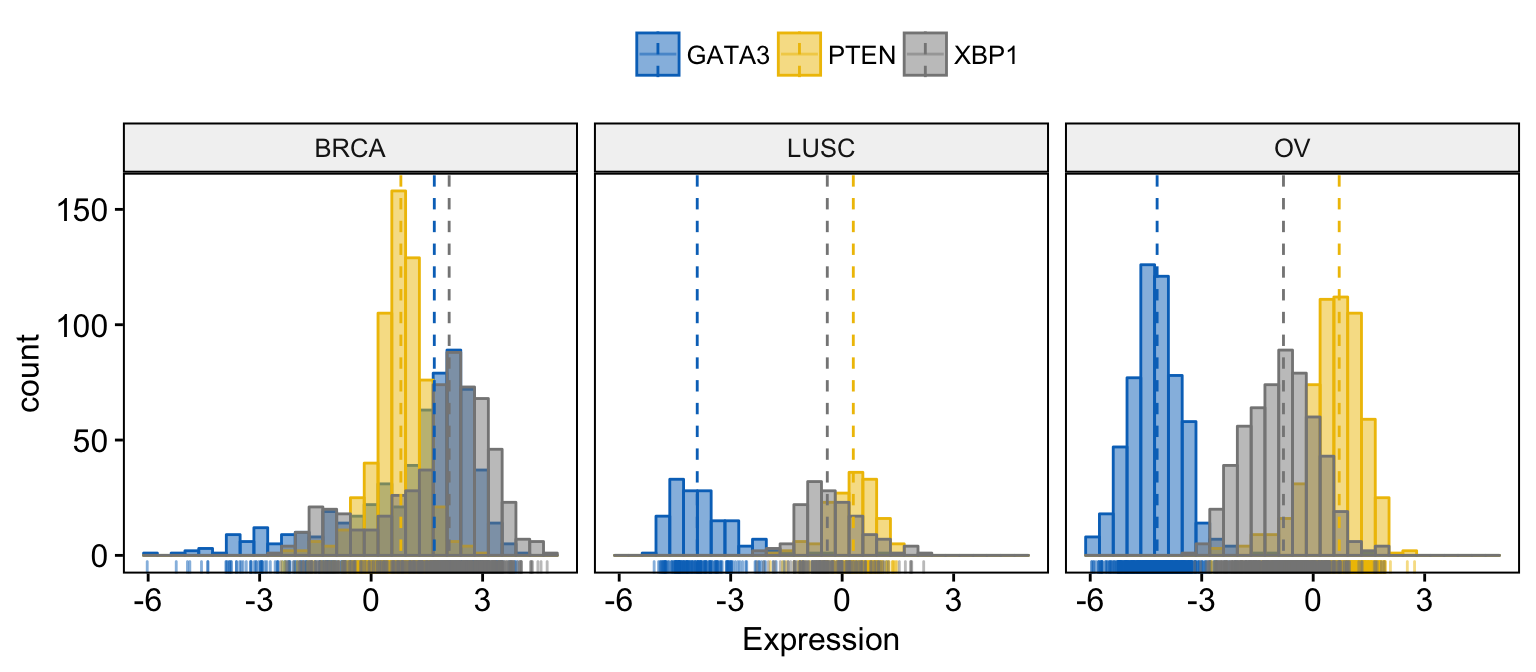
Exploratory Data visualization: Gene Expression Data
Empirical cumulative density function
(ggplot2 way of creating ECDF plots)
# Basic ECDF plot
ggecdf(expr,
x = c("GATA3", "PTEN", "XBP1"),
combine = TRUE,
xlab = "Expression", ylab = "F(expression)"
)Clik here to view.
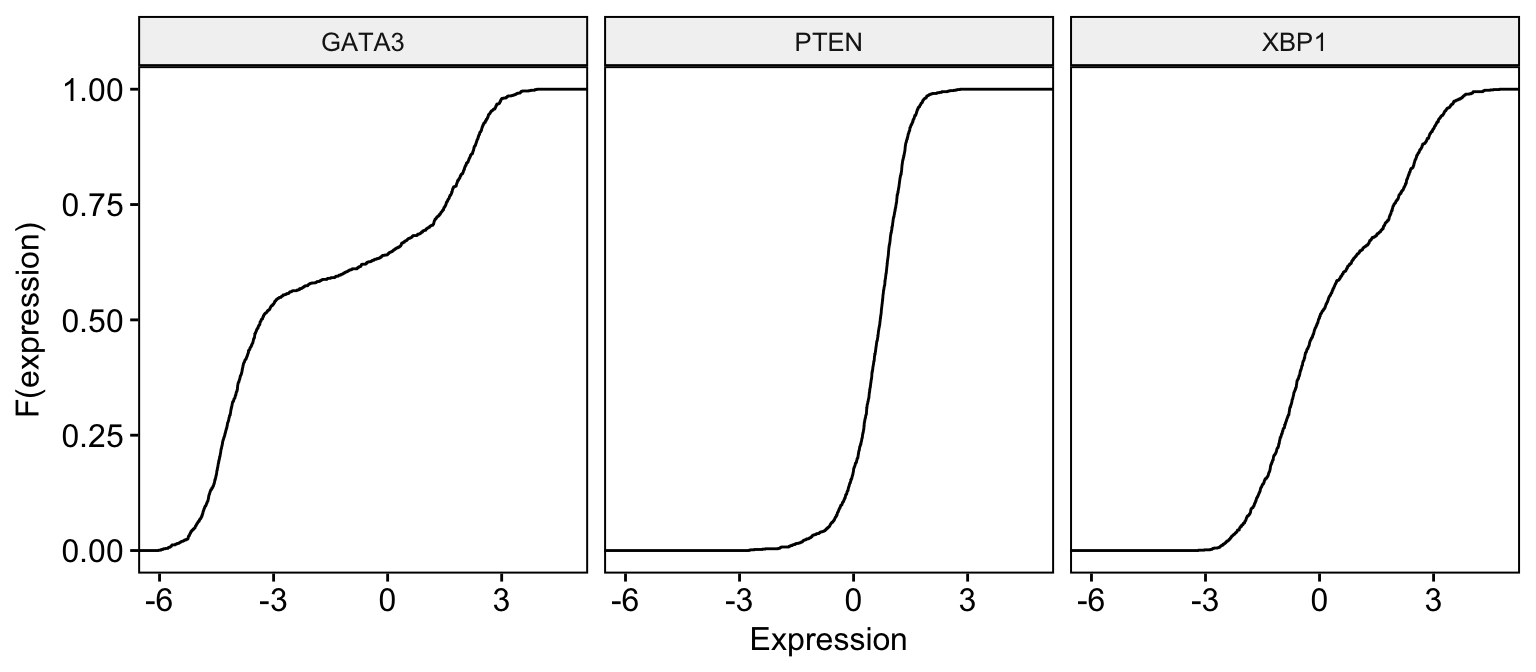
Exploratory Data visualization: Gene Expression Data
# Change color by dataset
ggecdf(expr,
x = c("GATA3", "PTEN", "XBP1"),
combine = TRUE,
xlab = "Expression", ylab = "F(expression)",
color = "dataset", palette = "jco"
)Clik here to view.
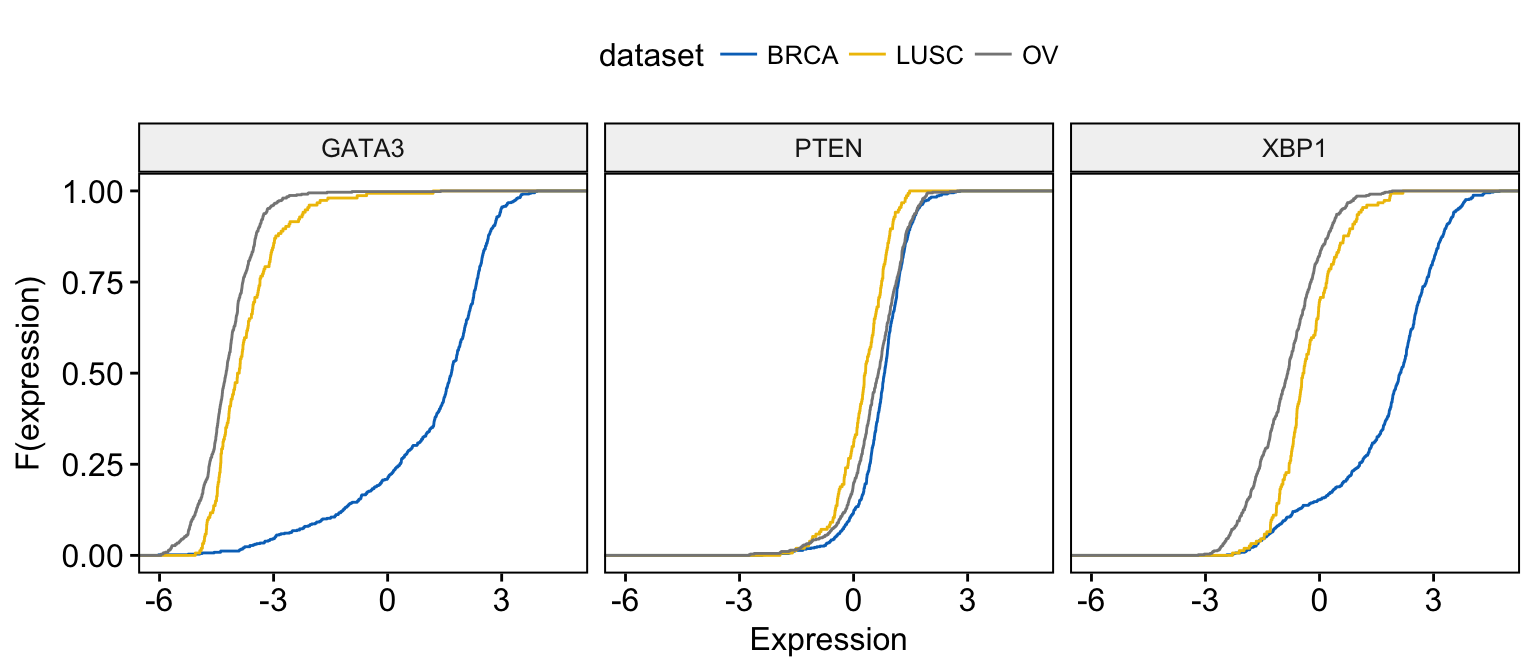
Exploratory Data visualization: Gene Expression Data
# Merge the 3 plots and color by x variables
ggecdf(expr,
x = c("GATA3", "PTEN", "XBP1"),
merge = TRUE,
xlab = "Expression", ylab = "F(expression)",
color = ".x.", palette = "jco"
)Clik here to view.
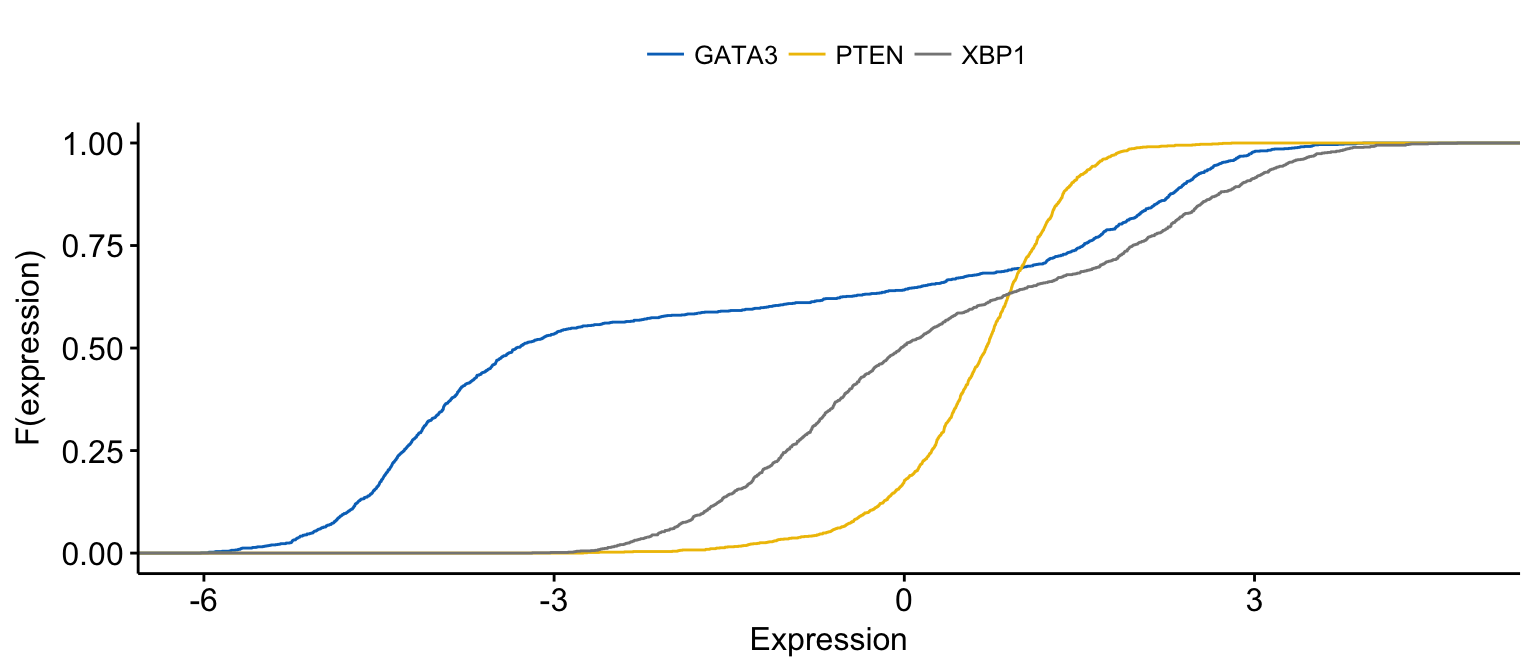
Exploratory Data visualization: Gene Expression Data
# Merge the 3 plots and color by x variables
# facet by "dataset" into multi-panel
ggecdf(expr,
x = c("GATA3", "PTEN", "XBP1"),
merge = TRUE,
xlab = "Expression", ylab = "F(expression)",
color = ".x.", palette = "jco",
facet.by = "dataset"
)Clik here to view.
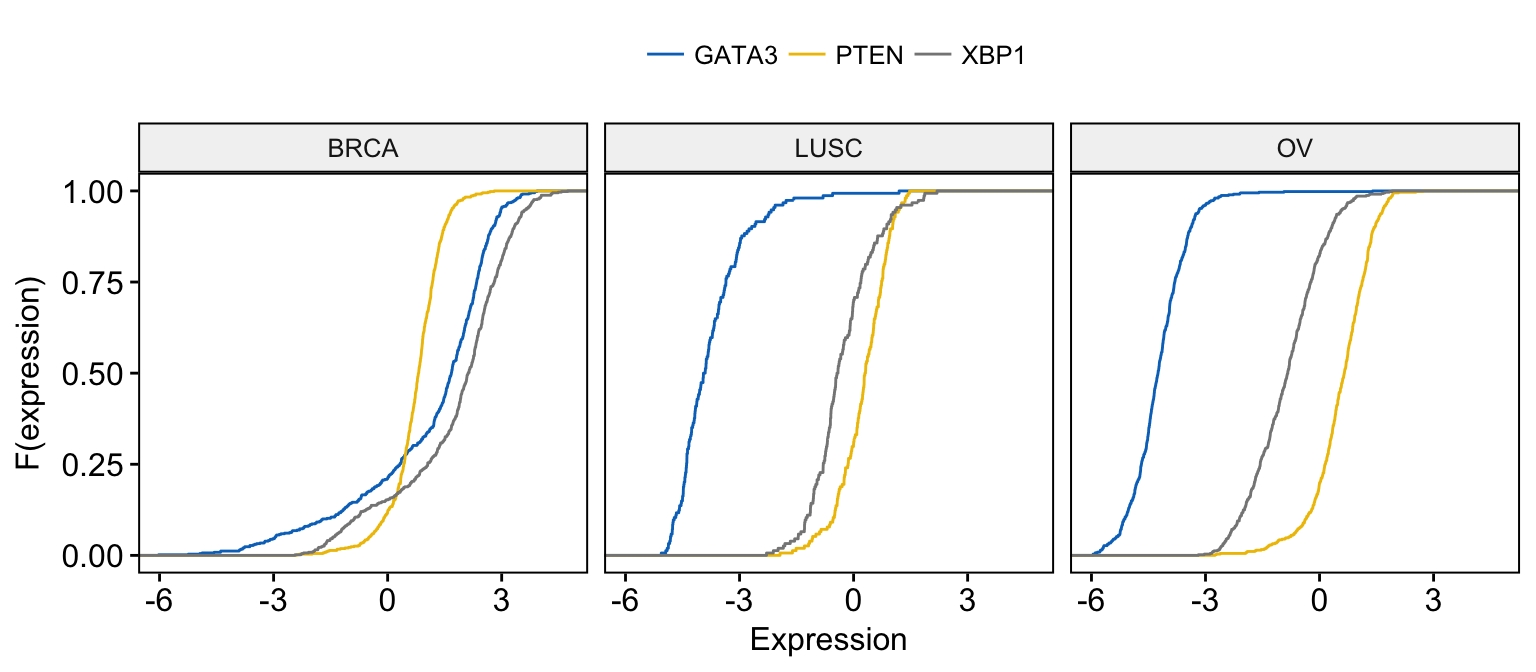
Exploratory Data visualization: Gene Expression Data
Quantile - Quantile plot
(ggplot2 way of creating QQ plots)
# Basic ECDF plot
ggqqplot(expr,
x = c("GATA3", "PTEN", "XBP1"),
combine = TRUE, size = 0.5
)Clik here to view.
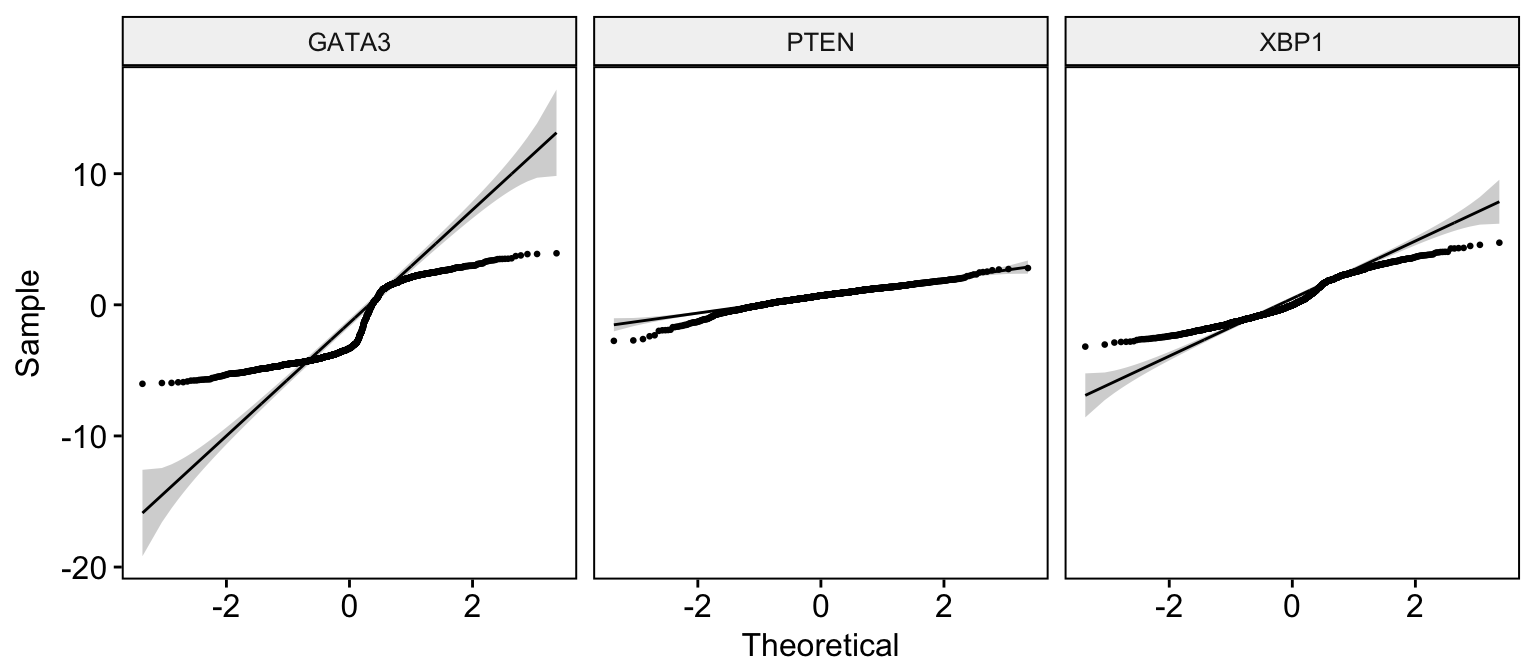
Exploratory Data visualization: Gene Expression Data
# Change color by dataset
ggqqplot(expr,
x = c("GATA3", "PTEN", "XBP1"),
combine = TRUE, color = "dataset", palette = "jco",
size = 0.5
)Clik here to view.
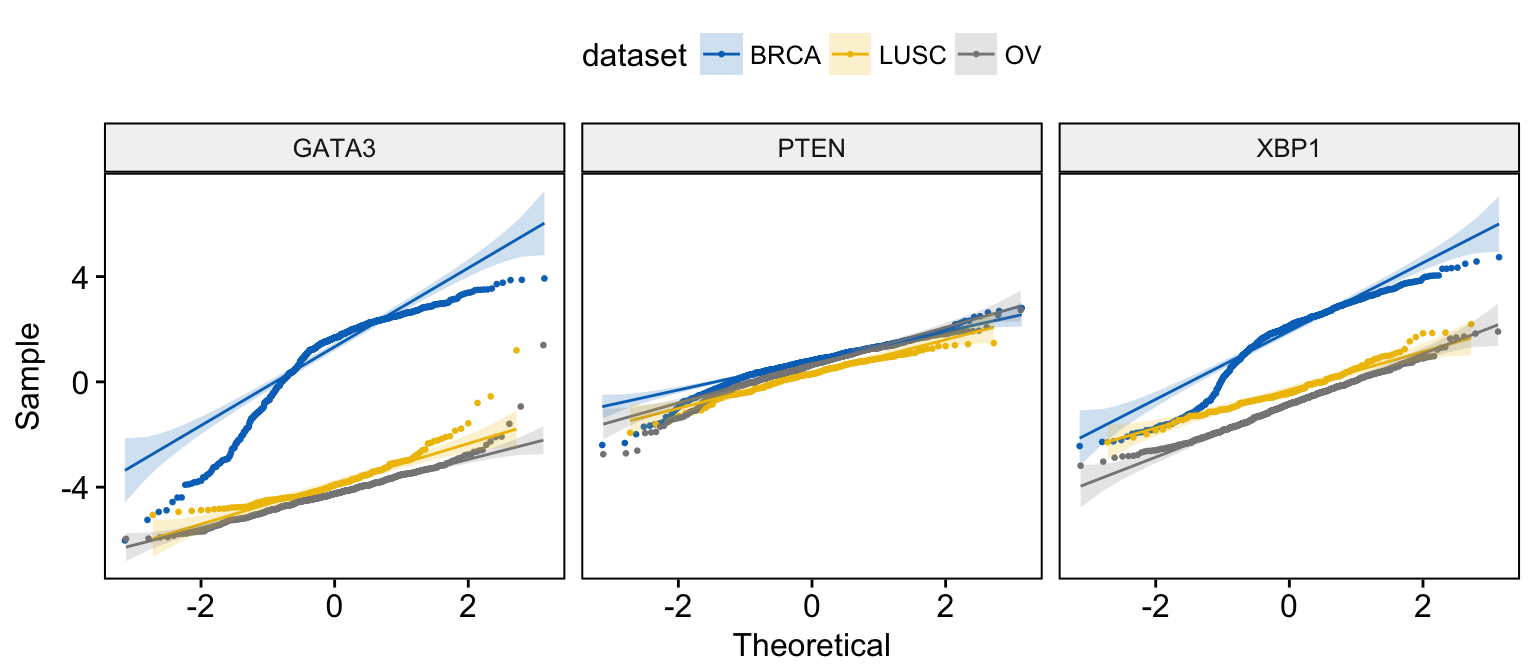
Exploratory Data visualization: Gene Expression Data
# Merge the 3 plots and color by x variables
ggqqplot(expr,
x = c("GATA3", "PTEN", "XBP1"),
merge = TRUE,
color = ".x.", palette = "jco"
)Clik here to view.
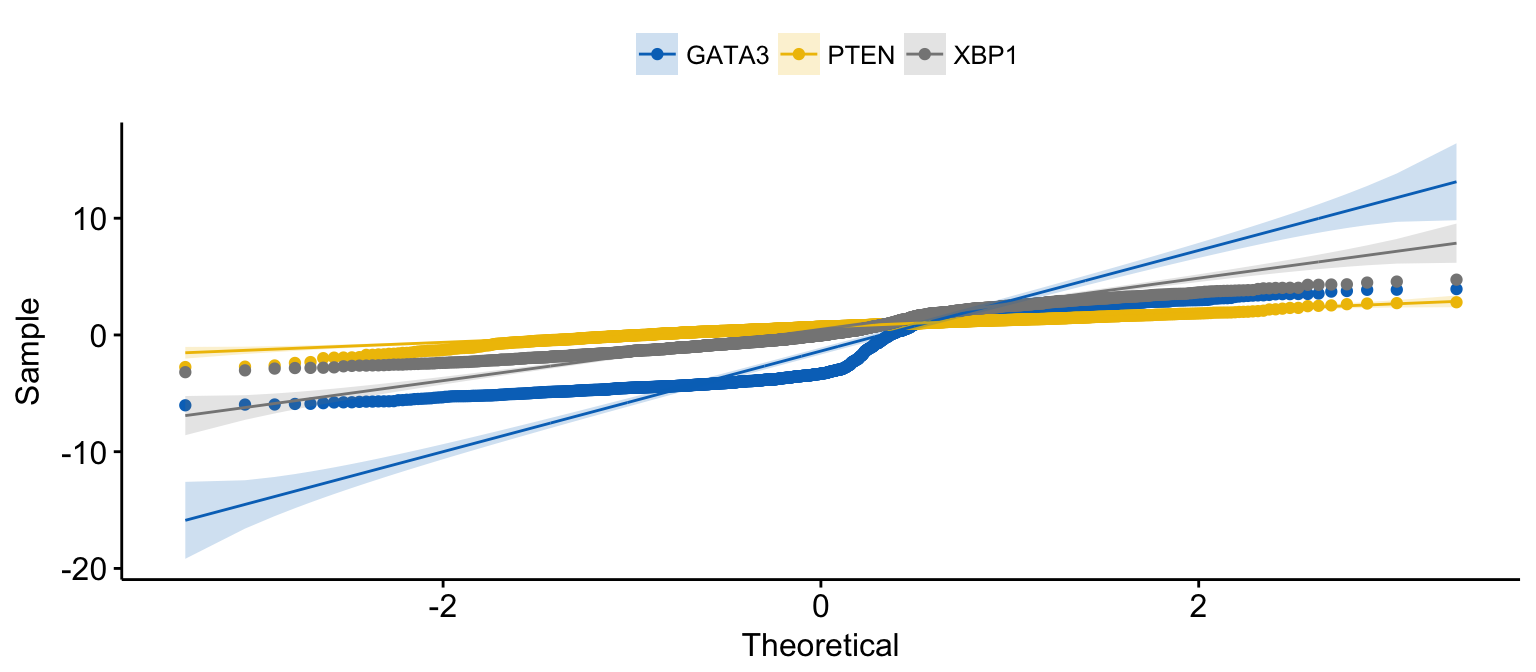
Exploratory Data visualization: Gene Expression Data
# Merge the 3 plots and color by x variables
# facet by "dataset" into multi-panel
ggqqplot(expr,
x = c("GATA3", "PTEN", "XBP1"),
merge = TRUE, size = 0.5,
color = ".x.", palette = "jco",
facet.by = "dataset"
)Clik here to view.
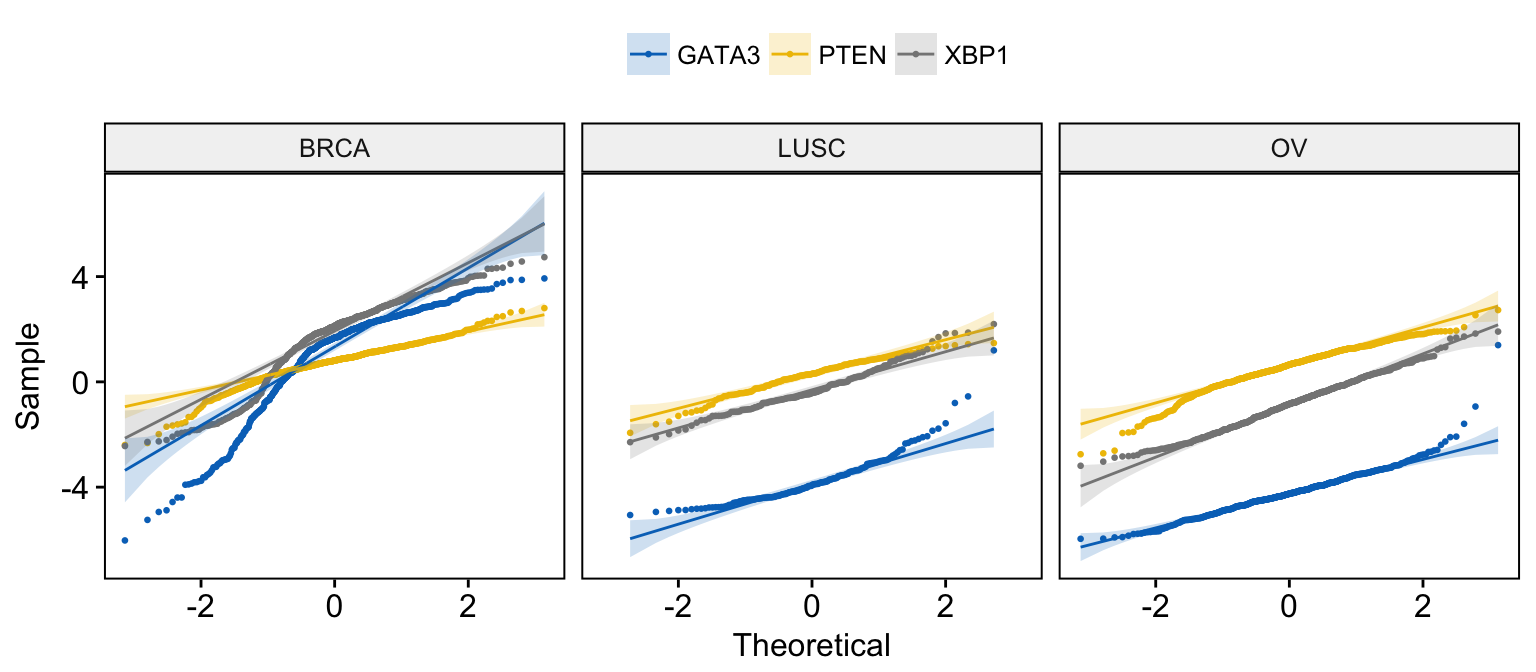
Exploratory Data visualization: Gene Expression Data
Infos
This analysis has been performed using R software (ver. 3.3.2) and ggpubr (ver. 0.1.3).